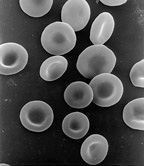
Red blood cells (RBCs), scientific name erythrocytes (from Greek erythros 'red' and kytos 'hollow vessel', with -cyte translated as 'cell' in modern usage), also referred to as red cells, red blood corpuscles (in humans or other animals not having nucleus in red blood cells) or haematids, are the most common type of blood cell and the vertebrate's principal means of delivering oxygen (O2) to the body tissues—via blood flow through the circulatory system. Erythrocytes take up oxygen in the lungs, or in fish the gills, and release it into tissues while squeezing through the body's capillaries.

Hemolysis or haemolysis, also known by several other names, is the rupturing (lysis) of red blood cells (erythrocytes) and the release of their contents (cytoplasm) into surrounding fluid. Hemolysis may occur in vivo or in vitro.

Spherocytosis is the presence of spherocytes in the blood, i.e. erythrocytes that are sphere-shaped rather than bi-concave disk shaped as normal. Spherocytes are found in all hemolytic anemias to some degree. Hereditary spherocytosis and autoimmune hemolytic anemia are characterized by having only spherocytes.
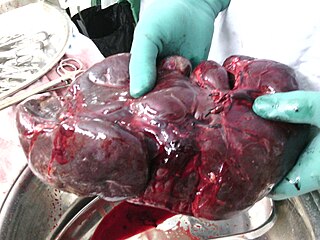
A splenectomy is the surgical procedure that partially or completely removes the spleen. The spleen is an important organ in regard to immunological function due to its ability to efficiently destroy encapsulated bacteria. Therefore, removal of the spleen runs the risk of overwhelming post-splenectomy infection, a medical emergency and rapidly fatal disease caused by the inability of the body's immune system to properly fight infection following splenectomy or asplenia.

Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired, life-threatening disease of the blood characterized by destruction of red blood cells by the complement system, a part of the body's innate immune system. This destructive process occurs due to deficiency of the red blood cell surface protein DAF, which normally inhibits such immune reactions. Since the complement cascade attacks the red blood cells within the blood vessels of the circulatory system, the red blood cell destruction (hemolysis) is considered an intravascular hemolytic anemia. There is ongoing research into other key features of the disease, such as the high incidence of venous blood clot formation. Research suggests that PNH thrombosis is caused by both the absence of GPI-anchored complement regulatory proteins on PNH platelets and the excessive consumption of nitric oxide (NO).

Hemolytic anemia or haemolytic anaemia is a form of anemia due to hemolysis, the abnormal breakdown of red blood cells (RBCs), either in the blood vessels or elsewhere in the human body (extravascular). This most commonly occurs within the spleen, but also can occur in the reticuloendothelial system or mechanically. Hemolytic anemia accounts for 5% of all existing anemias. It has numerous possible consequences, ranging from general symptoms to life-threatening systemic effects. The general classification of hemolytic anemia is either intrinsic or extrinsic. Treatment depends on the type and cause of the hemolytic anemia.
An autosplenectomy is a negative outcome of disease and occurs when a disease damages the spleen to such an extent that it becomes shrunken and non-functional. The spleen is an important immunological organ that acts as a filter for red blood cells, triggers phagocytosis of invaders, and mounts an immunological response when necessary. Lack of a spleen, called asplenia, can occur by autosplenectomy or the surgical counterpart, splenectomy. Asplenia can increase susceptibility to infection. Autosplenectomy can occur in cases of sickle-cell disease where the misshapen cells block blood flow to the spleen, causing scarring and eventual atrophy of the organ. Autosplenectomy is a rare condition that is linked to certain diseases but is not a common occurrence. It is also seen in systemic lupus erythematosus (SLE).
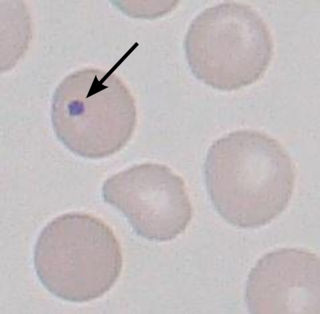
A Howell–Jolly body is a cytopathological finding of basophilic nuclear remnants in circulating erythrocytes. During maturation in the bone marrow, late erythroblasts normally expel their nuclei; but, in some cases, a small portion of DNA remains. The presence of Howell–Jolly bodies usually signifies a damaged or absent spleen, because a healthy spleen would normally filter such erythrocytes.
Warm antibody autoimmune hemolytic anemia (WAIHA) is the most common form of autoimmune haemolytic anemia. About half of the cases are of unknown cause, with the other half attributable to a predisposing condition or medications being taken. Contrary to cold autoimmune hemolytic anemia which happens in cold temperature (28–31 °C), WAIHA happens at body temperature.

Codocytes, also known as target cells, are red blood cells that have the appearance of a shooting target with a bullseye. In optical microscopy these cells appear to have a dark center surrounded by a white ring, followed by dark outer (peripheral) second ring containing a band of hemoglobin. However, in electron microscopy they appear very thin and bell shaped. Because of their thinness they are referred to as leptocytes. On routine smear morphology, some people like to make a distinction between leptocytes and codocytes- suggesting that in leptocytes the central spot is not completely detached from the peripheral ring, i.e. the pallor is in a C shape rather than a full ring.

Hereditary elliptocytosis, also known as ovalocytosis, is an inherited blood disorder in which an abnormally large number of the person's red blood cells are elliptical rather than the typical biconcave disc shape. Such morphologically distinctive erythrocytes are sometimes referred to as elliptocytes or ovalocytes. It is one of many red-cell membrane defects. In its severe forms, this disorder predisposes to haemolytic anaemia. Although pathological in humans, elliptocytosis is normal in camelids.

Hereditary stomatocytosis describes a number of inherited, mostly autosomal dominant human conditions which affect the red blood cell and create the appearance of a slit-like area of central pallor (stomatocyte) among erythrocytes on peripheral blood smear. The erythrocytes' cell membranes may abnormally 'leak' sodium and/or potassium ions, causing abnormalities in cell volume. Hereditary stomatocytosis should be distinguished from acquired causes of stomatocytosis, including dilantin toxicity and alcoholism, as well as artifact from the process of preparing peripheral blood smears.

Hereditary pyropoikilocytosis (HPP) is an autosomal recessive form of hemolytic anemia characterized by an abnormal sensitivity of red blood cells to heat and erythrocyte morphology similar to that seen in thermal burns or from prolonged exposure of a healthy patient's blood sample to high ambient temperatures. Patients with HPP tend to experience severe hemolysis and anemia in infancy that gradually improves, evolving toward typical elliptocytosis later in life. However, the hemolysis can lead to rapid sequestration and destruction of red cells. Splenectomy is curative when this occurs.
Autoimmune hemolytic anemia (AIHA) occurs when antibodies directed against the person's own red blood cells (RBCs) cause them to burst (lyse), leading to an insufficient number of oxygen-carrying red blood cells in the circulation. The lifetime of the RBCs is reduced from the normal 100–120 days to just a few days in serious cases. The intracellular components of the RBCs are released into the circulating blood and into tissues, leading to some of the characteristic symptoms of this condition. The antibodies are usually directed against high-incidence antigens, therefore they also commonly act on allogenic RBCs. AIHA is a relatively rare condition, with an incidence of 5–10 cases per 1 million persons per year in the warm-antibody type and 0.45 to 1.9 cases per 1 million persons per year in the cold antibody type. Autoimmune hemolysis might be a precursor of later onset systemic lupus erythematosus.
Normocytic anemia is a type of anemia and is a common issue that occurs for men and women typically over 85 years old. Its prevalence increases with age, reaching 44 percent in men older than 85 years. The most common type of normocytic anemia is anemia of chronic disease.

6-Phosphogluconate dehydrogenase deficiency, or partial deficiency, is an autosomal hereditary disease characterized by abnormally low levels of 6-phosphogluconate dehydrogenase (6PGD), a metabolic enzyme involved in the Pentose phosphate pathway. It is very important in the metabolism of red blood cells (erythrocytes). 6PDG deficiency affects less than 1% of the population, and studies suggest that there may be race variant involved in many of the reported cases. Although it is similar, 6PDG deficiency is not linked to glucose-6-phosphate dehydrogenase (G6PD) deficiency, as they are located on different chromosomes. However, a few people have had both of these metabolic diseases.
Congenital hemolytic anemia (CHA) is a diverse group of rare hereditary conditions marked by decreased life expectancy and premature removal of erythrocytes from blood flow. Defects in erythrocyte membrane proteins and red cell enzyme metabolism, as well as changes at the level of erythrocyte precursors, lead to impaired bone marrow erythropoiesis. CAH is distinguished by variable anemia, chronic extravascular hemolysis, decreased erythrocyte life span, splenomegaly, jaundice, biliary lithiasis, and iron overload. Immune-mediated mechanisms may play a role in the pathogenesis of these uncommon diseases, despite the paucity of data regarding the immune system's involvement in CHAs.
Erythrocyte fragility refers to the propensity of erythrocytes to hemolyse (rupture) under stress. It can be thought of as the degree or proportion of hemolysis that occurs when a sample of red blood cells are subjected to stress. Depending on the application as well as the kind of fragility involved, the amount of stress applied and/or the significance of the resultant hemolysis may vary.
Intravascular hemolysis describes hemolysis that happens mainly inside the vasculature. As a result, the contents of the red blood cell are released into the general circulation, leading to hemoglobinemia and increasing the risk of ensuing hyperbilirubinemia.
Hemolytic jaundice, also known as prehepatic jaundice, is a type of jaundice arising from hemolysis or excessive destruction of red blood cells, when the byproduct bilirubin is not excreted by the hepatic cells quickly enough. Unless the patient is concurrently affected by hepatic dysfunctions or is experiencing hepatocellular damage, the liver does not contribute to this type of jaundice.






