
Hydrogenation is a chemical reaction between molecular hydrogen (H2) and another compound or element, usually in the presence of a catalyst such as nickel, palladium or platinum. The process is commonly employed to reduce or saturate organic compounds. Hydrogenation typically constitutes the addition of pairs of hydrogen atoms to a molecule, often an alkene. Catalysts are required for the reaction to be usable; non-catalytic hydrogenation takes place only at very high temperatures. Hydrogenation reduces double and triple bonds in hydrocarbons.
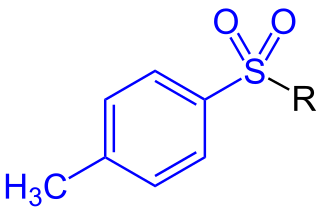
In organic chemistry, a toluenesulfonyl group (tosyl group, abbreviated Ts or Tos) is a univalent functional group with the chemical formula −SO2−C6H4−CH3. It consists of a tolyl group, −C6H4−CH3, joined to a sulfonyl group, −SO2−, with the open valence on sulfur. This group is usually derived from the compound tosyl chloride, CH3C6H4SO2Cl (abbreviated TsCl), which forms esters and amides of toluenesulfonic acid, CH3C6H4SO2OH (abbreviated TsOH). The para orientation illustrated (p-toluenesulfonyl) is most common, and by convention tosyl without a prefix refers to the p-toluenesulfonyl group.
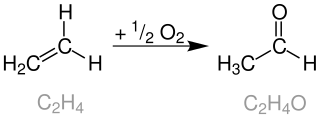
The Wacker process or the Hoechst-Wacker process refers to the oxidation of ethylene to acetaldehyde in the presence of palladium(II) chloride and copper(II) chloride as the catalyst. This chemical reaction was one of the first homogeneous catalysis with organopalladium chemistry applied on an industrial scale.
Clemmensen reduction is a chemical reaction described as a reduction of ketones or aldehydes to alkanes using zinc amalgam and concentrated hydrochloric acid (HCl). This reaction is named after Erik Christian Clemmensen, a Danish-American chemist.

The Pauson–Khand (PK) reaction is a chemical reaction, described as a [2+2+1] cycloaddition. In it, an alkyne, an alkene and carbon monoxide combine into a α,β-cyclopentenone in the presence of a metal-carbonyl catalyst.
The Negishi coupling is a widely employed transition metal catalyzed cross-coupling reaction. The reaction couples organic halides or triflates with organozinc compounds, forming carbon-carbon bonds (C-C) in the process. A palladium (0) species is generally utilized as the metal catalyst, though nickel is sometimes used. A variety of nickel catalysts in either Ni0 or NiII oxidation state can be employed in Negishi cross couplings such as Ni(PPh3)4, Ni(acac)2, Ni(COD)2 etc.
The Wurtz–Fittig reaction is the chemical reaction of aryl halides with alkyl halides and sodium metal in the presence of dry ether to give substituted aromatic compounds. Charles Adolphe Wurtz reported what is now known as the Wurtz reaction in 1855, involving the formation of a new carbon-carbon bond by coupling two alkyl halides. Work by Wilhelm Rudolph Fittig in the 1860s extended the approach to the coupling of an alkyl halide with an aryl halide. This modification of the Wurtz reaction is considered a separate process and is named for both scientists.
A carbometallation is any reaction where a carbon-metal bond reacts with a carbon-carbon π-bond to produce a new carbon-carbon σ-bond and a carbon-metal σ-bond. The resulting carbon-metal bond can undergo further carbometallation reactions or it can be reacted with a variety of electrophiles including halogenating reagents, carbonyls, oxygen, and inorganic salts to produce different organometallic reagents. Carbometallations can be performed on alkynes and alkenes to form products with high geometric purity or enantioselectivity, respectively. Some metals prefer to give the anti-addition product with high selectivity and some yield the syn-addition product. The outcome of syn and anti- addition products is determined by the mechanism of the carbometallation.
The Fukuyama coupling is a coupling reaction taking place between a thioester and an organozinc halide in the presence of a palladium catalyst. The reaction product is a ketone. This reaction was discovered by Tohru Fukuyama et al. in 1998.

The Achmatowicz reaction, also known as the Achmatowicz rearrangement, is an organic synthesis in which a furan is converted to a dihydropyran. In the original publication by the Polish Chemist Osman Achmatowicz Jr. in 1971 furfuryl alcohol is reacted with bromine in methanol to 2,5-dimethoxy-2,5-dihydrofuran which rearranges to the dihydropyran with dilute sulfuric acid. Additional reaction steps, alcohol protection with methyl orthoformate and boron trifluoride) and then ketone reduction with sodium borohydride produce an intermediate from which many monosaccharides can be synthesised.
Chiral Lewis acids (CLAs) are a type of Lewis acid catalyst. These acids affect the chirality of the substrate as they react with it. In such reactions, synthesis favors the formation of a specific enantiomer or diastereomer. The method is an enantioselective asymmetric synthesis reaction. Since they affect chirality, they produce optically active products from optically inactive or mixed starting materials. This type of preferential formation of one enantiomer or diastereomer over the other is formally known as asymmetric induction. In this kind of Lewis acid, the electron-accepting atom is typically a metal, such as indium, zinc, lithium, aluminium, titanium, or boron. The chiral-altering ligands employed for synthesizing these acids often have multiple Lewis basic sites that allow the formation of a ring structure involving the metal atom.

Basketane is a polycyclic alkane with the chemical formula C10H12. The name is taken from its structural similarity to a basket shape. Basketane was first synthesized in 1966, independently by Masamune and Dauben and Whalen. A patent application published in 1988 used basketane, which is a hydrocarbon, as a source material in doping thin diamond layers because of the molecule's high vapor pressure, carbon ring structure, and fewer hydrogen-to-carbon bond ratio.
David Markham Lemal is the Albert W. Smith Professor of Chemistry Emeritus and Research Professor of Chemistry at Dartmouth College. He received an A.B. degree (summa) from Amherst College in 1955 and a Ph.D. in chemistry from Harvard University in 1959. At Harvard he worked with R. B. Woodward on deoxy sugars and a synthesis of the alkaloid yohimbine.
In organic chemistry, the Fujiwara–Moritani reaction is a type of cross coupling reaction where an aromatic C-H bond is directly coupled to an olefinic C-H bond, generating a new C-C bond. This reaction is performed in the presence of a transition metal, typically palladium. The reaction was discovered by Yuzo Fujiwara and Ichiro Moritani in 1967. An external oxidant is required to this reaction to be run catalytically. Thus, this reaction can be classified as a C-H activation reaction, an oxidative Heck reaction, and a C-H olefination. Surprisingly, the Fujiwara–Moritani reaction was discovered before the Heck reaction.
The Riley oxidation is a selenium dioxide-mediated oxidation of methylene groups adjacent to carbonyls. It was first reported by Riley and co-workers in 1932. In the decade that ensued, selenium-mediated oxidation rapidly expanded in use, and in 1939, Guillemonat and co-workers disclosed the selenium dioxide-mediated oxidation of olefins at the allylic position. Today, selenium-dioxide-mediated oxidation of methylene groups to alpha ketones and at the allylic position of olefins is known as the Riley Oxidation.
Clark Landis is an American chemist, whose research focuses on organic and inorganic chemistry. He is currently a Professor of Chemistry at the University of Wisconsin–Madison. He was awarded the ACS Award in Organometallic Chemistry in 2010, and is a fellow of the American Chemical Society and the American Association for the Advancement of Science.
A phosphetane is a 4-membered organophosphorus heterocycle. The parent phosphetane molecule, which has the formula C3H7P, is one atom larger than phosphiranes, one smaller than phospholes, and is the heavy-atom analogue of azetidines. The first known phosphetane synthesis was reported in 1957 by Kosolapoff and Struck, but the method was both inefficient and hard to reproduce, with yields rarely exceeding 1%. A far more efficient method was reported in 1962 by McBride, whose method allowed for the first studies into the physical and chemical properties of phosphetanes. Phosphetanes are a well understood class of molecules that have found broad applications as chemical building blocks, reagents for organic/inorganic synthesis, and ligands in coordination chemistry.

The Mizoroki−Heck coupling of aryl halides and alkenes to form C(sp2)–C(sp2) bonds has become a staple transformation in organic synthesis, owing to its broad functional group compatibility and varied scope. In stark contrast, the palladium-catalyzed reductive Heck reaction has received considerably less attention, despite the fact that early reports of this reaction date back almost half a century. From the perspective of retrosynthetic logic, this transformation is highly enabling because it can forge alkyl–aryl linkages from widely available alkenes, rather than from the less accessible and/or more expensive alkyl halide or organometallic C(sp3) synthons that are needed in a classical aryl/alkyl cross-coupling.
T.V. (Babu) RajanBabu is an organic chemist who holds the position of Distinguished Professor of Chemistry in the College of Arts and Sciences at the Ohio State University. His laboratory traditionally focuses on developing transition metal-catalyzed reactions. RajanBabu is known for helping develop the Nugent-RajanBabu reagent, a chemical reagent used in synthetic organic chemistry as a single electron reductant.

The nitro-Mannich reaction is the nucleophilic addition of a nitroalkane to an imine, resulting in the formation of a beta-nitroamine. With the reaction involving the addition of an acidic carbon nucleophile to a carbon-heteroatom double bond, the nitro-Mannich reaction is related to some of the most fundamental carbon-carbon bond forming reactions in organic chemistry, including the aldol reaction, Henry reaction and Mannich reaction.