Related Research Articles
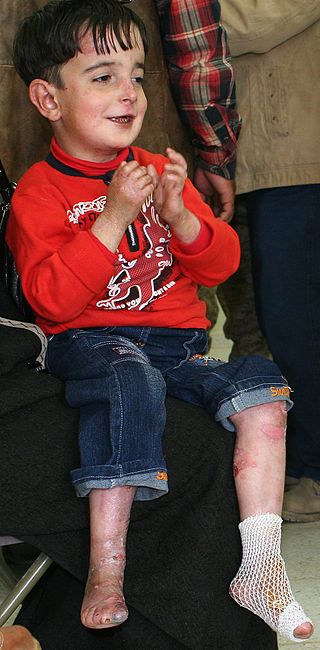
Epidermolysis bullosa (EB) is a group of rare medical conditions that result in easy blistering of the skin and mucous membranes. Blisters occur with minor trauma or friction and are painful. Its severity can range from mild to fatal. Inherited EB is a rare disease with a prevalence in the United States of 8.2 per million live births. Those with mild cases may not develop symptoms until they start to crawl or walk. Complications may include esophageal narrowing, squamous cell skin cancer, and the need for amputations.

Keratin 14 is a member of the type I keratin family of intermediate filament proteins. Keratin 14 was the first type I keratin sequence determined. Keratin 14 is also known as cytokeratin-14 (CK-14) or keratin-14 (KRT14). In humans it is encoded by the KRT14 gene.

Hemidesmosomes are very small stud-like structures found in keratinocytes of the epidermis of skin that attach to the extracellular matrix. They are similar in form to desmosomes when visualized by electron microscopy, however, desmosomes attach to adjacent cells. Hemidesmosomes are also comparable to focal adhesions, as they both attach cells to the extracellular matrix. Instead of desmogleins and desmocollins in the extracellular space, hemidesmosomes utilize integrins. Hemidesmosomes are found in epithelial cells connecting the basal epithelial cells to the lamina lucida, which is part of the basal lamina. Hemidesmosomes are also involved in signaling pathways, such as keratinocyte migration or carcinoma cell intrusion.
Adams–Oliver syndrome (AOS) is a rare congenital disorder characterized by defects of the scalp and cranium, transverse defects of the limbs, and mottling of the skin.
Aplasia is a birth defect where an organ or tissue is wholly or largely absent. It is caused by a defect in a developmental process.

Kindler syndrome is a rare congenital disease of the skin caused by a mutation in the KIND1 gene.

Keratin 5, also known as KRT5, K5, or CK5, is a protein that is encoded in humans by the KRT5 gene. It dimerizes with keratin 14 and forms the intermediate filaments (IF) that make up the cytoskeleton of basal epithelial cells. This protein is involved in several diseases including epidermolysis bullosa simplex and breast and lung cancers.

Collagen XVII, previously called BP180, is a transmembrane protein which plays a critical role in maintaining the linkage between the intracellular and the extracellular structural elements involved in epidermal adhesion, identified by Diaz and colleagues in 1990.

Epidermolysis bullosa dystrophica or dystrophic EB (DEB) is an inherited disease affecting the skin and other organs.

Genodermatosis is a hereditary skin disease with three inherited modes including single gene inheritance, multiple gene inheritance and chromosome inheritance. There are many different types of genodermatosis; the prevalence of genodermatosis ranges from 1 per 6000 people to 1 per 500,000 people. Genodermatosis has influence on the texture, color and structure of skin cuticle and connective tissue, specific lesion site and clinical manifestations on the body vary depending on the type. In the spite of the variety and complexity of genodermatosis, there are still some common methods that can help people diagnose. After diagnosis, different types of genodermatosis require different levels of therapy including interventions, nursing interventions and treatments. Among that, research of therapy for some new, complex and rare types are still in the developing stage. The impact of genodermatosis not only can be seen in body but also can be seen in all aspects of patients' life, including but not limited to psychological, family life, economic conditions and social activities. Accordingly, the patients need treatment, support and help in these areas.

Collagen alpha-1(VII) chain is a protein that in humans is encoded by the COL7A1 gene. It is composed of a triple helical, collagenous domain flanked by two non-collagenous domains, and functions as an anchoring fibril between the dermal-epidermal junction in the basement membrane. Mutations in COL7A1 cause all types of dystrophic epidermolysis bullosa, and the exact mutations vary based on the specific type or subtype. It has been shown that interactions between the NC-1 domain of collagen VII and several other proteins, including laminin-5 and collagen IV, contribute greatly to the overall stability of the basement membrane.
Anchoring fibrils extend from the basal lamina of epithelial cells and attach to the lamina reticularis by wrapping around the reticular fiber bundles. The basal lamina and lamina reticularis together make up the basement membrane. Anchoring fibrils are essential to the functional integrity of the dermoepidermal junction.
Epidermolysis bullosa acquisita, also known as acquired epidermolysis bullosa, is a longterm autoimmune blistering skin disease. It generally presents with fragile skin that blisters and becomes red with or without trauma. Marked scarring is left with thin skin, milia and nail changes. It typically begins around age 50.

Aplasia cutis congenita is a rare disorder characterized by congenital absence of skin. Ilona J. Frieden classified ACC in 1986 into 9 groups on the basis of location of the lesions and associated congenital anomalies. The scalp is the most commonly involved area with lesser involvement of trunk and extremities. Frieden classified ACC with fetus papyraceus as type 5. This type presents as truncal ACC with symmetrical absence of skin in stellate or butterfly pattern with or without involvement of proximal limbs. It is the most common congenital cicatricial alopecia, and is a congenital focal absence of epidermis with or without evidence of other layers of the skin.
Junctional epidermolysis bullosa is a skin condition characterized by blister formation within the lamina lucida of the basement membrane zone.
Wrinkly skin syndrome(WSS) is a rare genetic condition characterized by sagging, wrinkled skin, low skin elasticity, and delayed fontanelle (soft spot) closure, along with a range of other symptoms. The disorder exhibits an autosomal recessive inheritance pattern with mutations in the ATP6V0A2 gene, leading to abnormal glycosylation events. There are only about 30 known cases of WSS as of 2010. Given its rarity and symptom overlap with other dermatological conditions, reaching an accurate diagnosis is difficult and requires specialized dermatological testing. Limited treatment options are available but long-term prognosis is variable from patient to patient, based on individual case studies. Some skin symptoms recede with increasing age, while progressive neurological advancement of the disorder causes seizures and mental deterioration later in life for some patients.
Focal facial dermal dysplasia is a rare genetically heterogeneous group of disorders that are characterized by congenital bilateral scar like facial lesions, with or without associated facial anomalies. It is characterized by hairless lesions with fingerprint like puckering of the skin, especially at the temples, due to alternating bands of dermal and epidermal atrophy.
Aplasia cutis-myopia syndrome is a rare genetic disorder characterized by a combination of aplasia cutis congenita, high myopia, and dysfunction of the cone-rods. Other findings include congenital nystagmus, atrophy of the iris and pigment epithelium, easily scarred skin and keratoconus. Only 4 cases have been described in medical literature. Transmission is either autosomal dominant or autosomal recessive.
References
- ↑ Bart, Bruce (1966). "Congenital localized absence of skin and associated abnormalities resembling epidermolysis bullosa. A new syndrome". Archives of Dermatology. 93 (3): 296–304. doi:10.1001/archderm.1966.01600210032005. PMID 5910871.
- ↑ Frieden, IJ (1986). "Aplasia cutis congenita: A clinical review and proposal for classification". Journal of the American Academy of Dermatology. 14 (4): 646–660. doi:10.1016/S0190-9622(86)70082-0. PMID 3514708.
- ↑ Christiano AM, Bart BJ, Epstein EH Jr, Uitto J (1996). "Genetic basis of Bart's syndrome: a glycine substitution mutation in the type VII collagen gene". Journal of Investigative Dermatology. 106 (6): 1340–2. doi: 10.1111/1523-1747.ep12349293 . PMID 8752681.