
A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Tietz syndrome, also called Tietz albinism-deafness syndrome or albinism and deafness of Tietz, is an autosomal dominant congenital disorder characterized by deafness and leucism. It is caused by a mutation in the microphthalmia-associated transcription factor (MITF) gene. Tietz syndrome was first described in 1963 by Walter Tietz (1927–2003) a German Physician working in California.

Fukuyama congenital muscular dystrophy (FCMD) is a rare, autosomal recessive form of muscular dystrophy mainly described in Japan but also identified in Turkish and Ashkenazi Jewish patients; fifteen cases were first described on 1960 by Dr. Yukio Fukuyama.

Fuchs dystrophy, also referred to as Fuchs endothelial corneal dystrophy (FECD) and Fuchs endothelial dystrophy (FED), is a slowly progressing corneal dystrophy that usually affects both eyes and is slightly more common in women than in men. Although early signs of Fuchs dystrophy are sometimes seen in people in their 30s and 40s, the disease rarely affects vision until people reach their 50s and 60s.

Corneal dystrophy is a group of rare hereditary disorders characterised by bilateral abnormal deposition of substances in the transparent front part of the eye called the cornea.
Autosomal recessive multiple epiphyseal dysplasia (ARMED), also called epiphyseal dysplasia, multiple, 4 (EDM4), multiple epiphyseal dysplasia with clubfoot or –with bilayered patellae, is an autosomal recessive congenital disorder affecting cartilage and bone development. The disorder has relatively mild signs and symptoms, including joint pain, scoliosis, and malformations of the hands, feet, and knees.
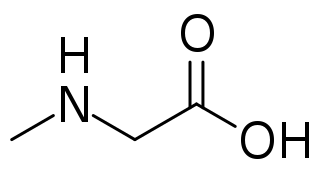
Sarcosinemia (SAR), also called hypersarcosinemia and SARDH deficiency, is a rare autosomal recessive metabolic disorder characterized by an increased concentration of sarcosine in blood plasma and urine ("sarcosinuria"). It can result from an inborn error of sarcosine metabolism, or from severe folate deficiency related to the folate requirement for the conversion of sarcosine to glycine. It is thought to be a relatively benign condition.

Axenfeld–Rieger syndrome is a rare autosomal dominant disorder, which affects the development of the teeth, eyes, and abdominal region.
White sponge nevus (WSN) is an autosomal dominant condition of the oral mucosa. It is caused by a mutations in certain genes coding for keratin, which causes a defect in the normal process of keratinization of the mucosa. This results in lesions which are thick, white and velvety on the inside of the cheeks within the mouth. Usually, these lesions are present from birth or develop during childhood. The condition is entirely harmless, and no treatment is required.
Naegeli–Franceschetti–Jadassohn syndrome (NFJS), also known as chromatophore nevus of Naegeli and Naegeli syndrome, is a rare autosomal dominant form of ectodermal dysplasia, characterized by reticular skin pigmentation, diminished function of the sweat glands, the absence of teeth and hyperkeratosis of the palms and soles. One of the most striking features is the absence of fingerprint lines on the fingers.

Bietti's crystalline dystrophy (BCD) is a rare autosomal recessive eye disease named after G. B. Bietti.
EEM syndrome is an autosomal recessive congenital malformation disorder affecting tissues associated with the ectoderm, and also the hands, feet and eyes.

Meesmann corneal dystrophy (MECD) is a rare hereditary autosomal dominant disease that is characterized as a type of corneal dystrophy and a keratin disease. MECD is characterized by the formation of microcysts in the outermost layer of the cornea, known as the anterior corneal epithelium. The anterior corneal epithelium also becomes fragile. This usually affects both eyes rather than a single eye and worsens over time. There are two phenotypes, Meesmann corneal dystrophy 1 (MECD1) and Meesmann corneal dystrophy 2 (MECD2), which affect the genes KRT3 and KRT12, respectively. A heterozygous mutation in either of these genes will lead to a single phenotype. Many with Meesmann corneal dystrophy are asymptomatic or experience mild symptoms.
Worth syndrome, also known as benign form of Worth hyperostosis corticalis generalisata with torus platinus, autosomal dominant osteosclerosis, autosomal dominant endosteal hyperostosis or Worth disease, is a rare autosomal dominant congenital disorder that is caused by a mutation in the LRP5 gene. It is characterized by increased bone density and benign bony structures on the palate.
Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.

Reis-Bücklers corneal dystrophy is a rare, corneal dystrophy of unknown cause, in which the Bowman's layer of the cornea undergoes disintegration. The disorder is inherited in an autosomal dominant fashion, and is associated with mutations in the gene TGFB1.

Granular corneal dystrophy is a slowly progressive corneal dystrophy that most often begins in early childhood.
Parastremmatic dwarfism is a rare bone disease that features severe dwarfism, thoracic kyphosis, a distortion and twisting of the limbs, contractures of the large joints, malformations of the vertebrae and pelvis, and incontinence. The disease was first reported in 1970 by Leonard Langer and associates; they used the term parastremmatic from the Greek parastremma, or distorted limbs, to describe it. On X-rays, the disease is distinguished by a "flocky" or lace-like appearance to the bones. The disease is congenital, which means it is apparent at birth. It is caused by a mutation in the TRPV4 gene, located on chromosome 12 in humans. The disease is inherited in an autosomal dominant manner.
Keratoendotheliitis fugax hereditaria is an autosomal dominantly inherited disease of the cornea, caused by a point mutation in cryopyrin that in humans is encoded by the NLRP3 gene located on the long arm of chromosome 1.

The human cornea is a transparent membrane which allows light to pass through it. The word corneal opacification literally means loss of normal transparency of cornea. The term corneal opacity is used particularly for the loss of transparency of cornea due to scarring. Transparency of the cornea is dependent on the uniform diameter and the regular spacing and arrangement of the collagen fibrils within the stroma. Alterations in the spacing of collagen fibrils in a variety of conditions including corneal edema, scars, and macular corneal dystrophy is clinically manifested as corneal opacity. The term "corneal blindness" is commonly used to describe blindness due to corneal opacity.

