Difficulty walking (ambulation is typically lost by age 5–15 years)
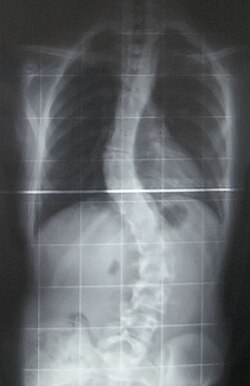
Contractures Characteristically, bilateral contractures of the proximal joints of the upper extremities (shoulder and elbows) and proximal joints of the lower extremities (hips and knees). Spine contractures in the form of progressive scoliosis occurs and occasionally contracture of neck musculature also known as torticollis.[10]
Joint looseness Contractures can be associated with distal joint laxity of the upper extremities (wrists and fingers) and of the lower extremities (ankle and toes).[10]
COL6A1 plays an important part in maintaining the human body's integrity of various tissues. Alpha 1 subunit of type VI collagen is the encoded protein.[11]
In terms of the diagnosis of Ullrich congenital muscular dystrophy upon inspection follicular hyperkeratosis, may be a dermatological indicator, additionally also serum creatine kinase may be mildly above normal.[6] Other exams/methods to ascertain if the individual has Ullrich congenital muscular dystrophy are:[medical citation needed]
Phenotypes of overlap between Ullrich congenital muscular dystrophy (UCMD) and Bethlem myopathy can be assumed. In the differential diagnosis of UCDM, even in patients without finger contractures, Bethlem myopathy could be considered.[13]
Treatment
Scoliosis X-ray
Treatment for Ullrich congenital muscular dystrophy can consist of physical therapy and regular stretching to prevent and reduce contractures. Respiratory support may be needed at some point by the affected individual.[3]
Though cardiac complications are not a concern in this type of CMD, in regards to respiratory issues ventilation via a tracheostomy is a possibility in some cases.[6][14]
Prognosis
The prognosis of this sub-type of MD indicates that the affected individual may eventually have feeding difficulties. Surgery, at some point, might be an option for scoliosis.[3]
Scoliosis, which is a sideways curve of the persons vertebrate, is determined by a variety of factors, including the degree (mild or severe), in which case if possible a brace might be used by the individual.[15]
Research
Cyclosporin-A
In terms of possible research for Ullrich congenital muscular dystrophy one source indicates that cyclosporine A might be of benefit to individuals with this CMD type.[16]
According to a review by Bernardi, et al., cyclosporin A (CsA) used to treat collagen VI muscular dystrophies demonstrates a normalization of mitochondrial reaction to rotenone.[17]
1 2 3 Foley, A. Reghan; Mohassel, Payam; Donkervoort, Sandra; Bolduc, Véronique; Bönnemann, Carsten G. (January 31, 1993). "Collagen VI-Related Dystrophies". In Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E.; Bean, Lora JH; Gripp, Karen W.; Mirzaa, Ghayda M.; Amemiya, Anne (eds.). GeneReviews®. University of Washington, Seattle. PMID20301676. Archived from the original on August 13, 2020. Retrieved November 11, 2012.
1 2 El-Sobky, Tamer A.; Abdulhady, Hala; Mahmoud, Shady; Amen, John (31 January 2024). "Orthopedic manifestations of congenital muscular dystrophy subtypes in children: Emerging signatures need consolidation: a scoping review". Journal of Musculoskeletal Surgery and Research. 8 11: 11–23. doi:10.25259/JMSR_229_2023.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.