
Mitochondrial disease is a group of disorders caused by mitochondrial dysfunction. Mitochondria are the organelles that generate energy for the cell and are found in every cell of the human body except red blood cells. They convert the energy of food molecules into the ATP that powers most cell functions.
Encephalopathy means any disorder or disease of the brain, especially chronic degenerative conditions. In modern usage, encephalopathy does not refer to a single disease, but rather to a syndrome of overall brain dysfunction; this syndrome has many possible organic and inorganic causes.
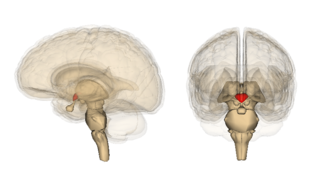
Wernicke encephalopathy (WE), also Wernicke's encephalopathy, or wet brain is the presence of neurological symptoms caused by biochemical lesions of the central nervous system after exhaustion of B-vitamin reserves, in particular thiamine (vitamin B1). The condition is part of a larger group of thiamine deficiency disorders that includes beriberi, in all its forms, and alcoholic Korsakoff syndrome. When it occurs simultaneously with alcoholic Korsakoff syndrome it is known as Wernicke–Korsakoff syndrome.

Wolfram syndrome, also called DIDMOAD, is a rare autosomal-recessive genetic disorder that causes childhood-onset diabetes mellitus, optic atrophy, and deafness as well as various other possible disorders including neurodegeneration. Symptoms can start to appear as early as childhood to adult years. There is a 25% recurrence risk in children.

Leigh syndrome is an inherited neurometabolic disorder that affects the central nervous system. It is named after Archibald Denis Leigh, a British neuropsychiatrist who first described the condition in 1951. Normal levels of thiamine, thiamine monophosphate, and thiamine diphosphate are commonly found, but there is a reduced or absent level of thiamine triphosphate. This is thought to be caused by a blockage in the enzyme thiamine-diphosphate kinase, and therefore treatment in some patients would be to take thiamine triphosphate daily. While the majority of patients typically exhibit symptoms between the ages of 3 and 12 months, instances of adult onset have also been documented.

Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.
Kearns–Sayre syndrome (KSS), oculocraniosomatic disorder or oculocranionsomatic neuromuscular disorder with ragged red fibers is a mitochondrial myopathy with a typical onset before 20 years of age. KSS is a more severe syndromic variant of chronic progressive external ophthalmoplegia, a syndrome that is characterized by isolated involvement of the muscles controlling movement of the eyelid and eye. This results in ptosis and ophthalmoplegia respectively. KSS involves a combination of the already described CPEO as well as pigmentary retinopathy in both eyes and cardiac conduction abnormalities. Other symptoms may include cerebellar ataxia, proximal muscle weakness, deafness, diabetes mellitus, growth hormone deficiency, hypoparathyroidism, and other endocrinopathies. In both of these diseases, muscle involvement may begin unilaterally but always develops into a bilateral deficit, and the course is progressive. This discussion is limited specifically to the more severe and systemically involved variant.

MELAS is one of the family of mitochondrial diseases, which also include MIDD, MERRF syndrome, and Leber's hereditary optic neuropathy. It was first characterized under this name in 1984. A feature of these diseases is that they are caused by defects in the mitochondrial genome which is inherited purely from the female parent. The most common MELAS mutation is mitochondrial mutation, mtDNA, referred to as m.3243A>G.

MERRF syndrome is a mitochondrial disease. It is extremely rare, and has varying degrees of expressivity owing to heteroplasmy. MERRF syndrome affects different parts of the body, particularly the muscles and nervous system. The signs and symptoms of this disorder appear at an early age, generally childhood or adolescence. The causes of MERRF syndrome are difficult to determine, but because it is a mitochondrial disorder, it can be caused by the mutation of nuclear DNA or mitochondrial DNA. The classification of this disease varies from patient to patient, since many individuals do not fall into one specific disease category. The primary features displayed on a person with MERRF include myoclonus, seizures, cerebellar ataxia, myopathy, and ragged red fibers (RRF) on muscle biopsy, leading to the disease's name. Secondary features include dementia, optic atrophy, bilateral deafness, peripheral neuropathy, spasticity, or multiple lipomata. Mitochondrial disorders, including MERRFS, may present at any age.

Purine nucleoside phosphorylase deficiency is a rare autosomal recessive metabolic disorder which results in immunodeficiency.
Pearson syndrome is a mitochondrial disease characterized by sideroblastic anemia and exocrine pancreas dysfunction. Other clinical features are failure to thrive, pancreatic fibrosis with insulin-dependent diabetes and exocrine pancreatic deficiency, muscle and neurologic impairment, and, frequently, early death. It is usually fatal in infancy. The few patients who survive into adulthood often develop symptoms of Kearns–Sayre syndrome. It is caused by a deletion in mitochondrial DNA. Pearson syndrome is very rare: fewer than a hundred cases have been reported in medical literature worldwide.
Chronic progressive external ophthalmoplegia (CPEO) is a type of eye disorder characterized by slowly progressive inability to move the eyes and eyebrows. It is often the only feature of mitochondrial disease, in which case the term CPEO may be given as the diagnosis. In other people suffering from mitochondrial disease, CPEO occurs as part of a syndrome involving more than one part of the body, such as Kearns–Sayre syndrome. Occasionally CPEO may be caused by conditions other than mitochondrial diseases.

TYMP is a gene that encodes for the enzyme thymidine phosphorylase. The TYMP gene is also known as ECGF1 and MNGIE due to its role in MNGIE syndrome.
Mitochondrially encoded tRNA histidine, also known as MT-TH, is a transfer RNA which, in humans, is encoded by the mitochondrial MT-TH gene.

DNA polymerase subunit gamma is an enzyme that in humans is encoded by the POLG gene. Mitochondrial DNA polymerase is heterotrimeric, consisting of a homodimer of accessory subunits plus a catalytic subunit. The protein encoded by this gene is the catalytic subunit of mitochondrial DNA polymerase. Defects in this gene are a cause of progressive external ophthalmoplegia with mitochondrial DNA deletions 1 (PEOA1), sensory ataxic neuropathy dysarthria and ophthalmoparesis (SANDO), Alpers-Huttenlocher syndrome (AHS), and mitochondrial neurogastrointestinal encephalopathy syndrome (MNGIE).

Thymidine phosphorylase is an enzyme that is encoded by the TYMP gene and catalyzes the reaction:

Twinkle protein also known as twinkle mtDNA helicase is a mitochondrial protein that in humans is encoded by the TWNK gene located in the long arm of chromosome 10 (10q24.31).
Mitochondrially encoded tRNA lysine also known as MT-TK is a transfer RNA which in humans is encoded by the mitochondrial MT-TK gene.
Mitochondrially encoded tRNA tyrosine, also known as MT-TY, is a transfer RNA which in humans is encoded by the mitochondrial MT-TY gene.
Mitochondrial DNA depletion syndrome, or Alper's disease, is any of a group of autosomal recessive disorders that cause a significant drop in mitochondrial DNA in affected tissues. Symptoms can be any combination of myopathic, hepatopathic, or encephalomyopathic. These syndromes affect tissue in the muscle, liver, or both the muscle and brain, respectively. The condition is typically fatal in infancy and early childhood, though some have survived to their teenage years with the myopathic variant and some have survived into adulthood with the SUCLA2 encephalomyopathic variant. There is currently no curative treatment for any form of MDDS, though some preliminary treatments have shown a reduction in symptoms.

