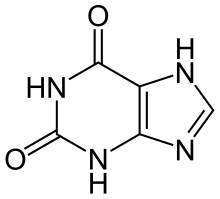
Uric acid is a heterocyclic compound of carbon, nitrogen, oxygen, and hydrogen with the formula C5H4N4O3. It forms ions and salts known as urates and acid urates, such as ammonium acid urate. Uric acid is a product of the metabolic breakdown of purine nucleotides, and it is a normal component of urine. High blood concentrations of uric acid can lead to gout and are associated with other medical conditions, including diabetes and the formation of ammonium acid urate kidney stones.

Xanthine is a purine base found in most human body tissues and fluids, as well as in other organisms. Several stimulants are derived from xanthine, including caffeine, theophylline, and theobromine.
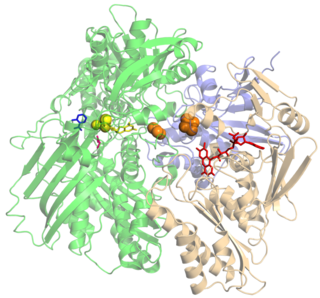
Xanthine oxidase is a form of xanthine oxidoreductase, a type of enzyme that generates reactive oxygen species. These enzymes catalyze the oxidation of hypoxanthine to xanthine and can further catalyze the oxidation of xanthine to uric acid. These enzymes play an important role in the catabolism of purines in some species, including humans.

Allopurinol is a medication used to decrease high blood uric acid levels. It is specifically used to prevent gout, prevent specific types of kidney stones and for the high uric acid levels that can occur with chemotherapy. It is taken orally or intravenously.
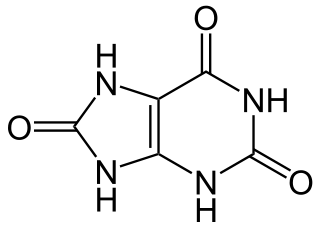
Hyperuricaemia or hyperuricemia is an abnormally high level of uric acid in the blood. In the pH conditions of body fluid, uric acid exists largely as urate, the ion form. Serum uric acid concentrations greater than 6 mg/dL for females, 7 mg/dL for males, and 5.5 mg/dL for youth are defined as hyperuricemia. The amount of urate in the body depends on the balance between the amount of purines eaten in food, the amount of urate synthesised within the body, and the amount of urate that is excreted in urine or through the gastrointestinal tract. Hyperuricemia may be the result of increased production of uric acid, decreased excretion of uric acid, or both increased production and reduced excretion.

Hypoxanthine-guanine phosphoribosyltransferase (HGPRT) is an enzyme encoded in humans by the HPRT1 gene.
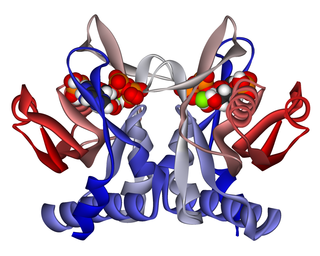
Adenine phosphoribosyltransferase (APRTase) is an enzyme encoded by the APRT gene, found in humans on chromosome 16. It is part of the Type I PRTase family and is involved in the nucleotide salvage pathway, which provides an alternative to nucleotide biosynthesis de novo in humans and most other animals. In parasitic protozoa such as giardia, APRTase provides the sole mechanism by which AMP can be produced. APRTase deficiency contributes to the formation of kidney stones (urolithiasis) and to potential kidney failure.

Hypouricemia or hypouricaemia is a level of uric acid in blood serum that is below normal. In humans, the normal range of this blood component has a lower threshold set variously in the range of 2 mg/dL to 4 mg/dL, while the upper threshold is 530 μmol/L (6 mg/dL) for women and 619 μmol/L (7 mg/dL) for men. Hypouricemia usually is benign and sometimes is a sign of a medical condition.

Adenine phosphoribosyltransferase deficiency is a rare autosomal recessive metabolic disorder caused by mutations of the APRT gene. Adenine phosphoribosyltransferase (APRT) catalyzes the creation of pyrophosphate and adenosine monophosphate from 5-phosphoribosyl-1-pyrophosphate and adenine. Adenine phosphoribosyltransferase is a purine salvage enzyme. Genetic mutations of adenine phosphoribosyltransferase make large amounts of 2,8-Dihydroxyadenine causing urolithiasis and renal failure.
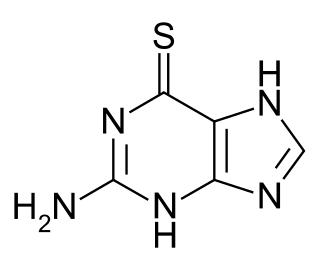
Tioguanine, also known as thioguanine or 6-thioguanine (6-TG) or tabloid is a medication used to treat acute myeloid leukemia (AML), acute lymphocytic leukemia (ALL), and chronic myeloid leukemia (CML). Long-term use is not recommended. It is given by mouth.

Nucleic acid metabolism is a collective term that refers to the variety of chemical reactions by which nucleic acids are either synthesized or degraded. Nucleic acids are polymers made up of a variety of monomers called nucleotides. Nucleotide synthesis is an anabolic mechanism generally involving the chemical reaction of phosphate, pentose sugar, and a nitrogenous base. Degradation of nucleic acids is a catabolic reaction and the resulting parts of the nucleotides or nucleobases can be salvaged to recreate new nucleotides. Both synthesis and degradation reactions require multiple enzymes to facilitate the event. Defects or deficiencies in these enzymes can lead to a variety of diseases.

Aldehyde oxidase (AO) is a metabolizing enzyme, located in the cytosolic compartment of tissues in many organisms. AO catalyzes the oxidation of aldehydes into carboxylic acid, and in addition, catalyzes the hydroxylation of some heterocycles. It can also catalyze the oxidation of both cytochrome P450 and monoamine oxidase (MAO) intermediate products. AO plays an important role in the metabolism of several drugs.

Purine nucleoside phosphorylase deficiency is a rare autosomal recessive metabolic disorder which results in immunodeficiency.
Purine metabolism refers to the metabolic pathways to synthesize and break down purines that are present in many organisms.

The human gene SRD5A2 encodes the 3-oxo-5α-steroid 4-dehydrogenase 2 enzyme, also known as 5α-reductase type 2 (5αR2), one of three isozymes of 5α-reductase.

Xanthine dehydrogenase, also known as XDH, is a protein that, in humans, is encoded by the XDH gene.

Molybdenum cofactor sulfurase is an enzyme that in humans is encoded by the MOCOS gene.
Molybdenum cofactor deficiency is a rare human disease in which the absence of molybdopterin – and consequently its molybdenum complex, commonly called molybdenum cofactor – leads to accumulation of toxic levels of sulphite and neurological damage. Usually this leads to death within months of birth, due to the lack of active sulfite oxidase. Furthermore, a mutational block in molybdenum cofactor biosynthesis causes absence of enzyme activity of xanthine dehydrogenase/oxidase and aldehyde oxidase.
Aldehyde oxidase 1 is an enzyme that in humans is encoded by the AOX1 gene.
Caffeine dehydrogenase, commonly referred to in scientific literature as caffeine oxidase, is an enzyme with the systematic name caffeine:ubiquinone oxidoreductase. The enzyme is most well known for its ability to directly oxidize caffeine, a type of methylxanthine, to trimethyluric acid. Caffeine dehydrogenase can be found in bacterium Pseudomonas sp. CBB1 and in several species within the genera Alcaligenes, Rhodococcus, and Klebsiella.