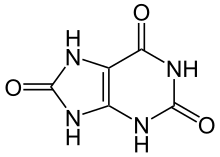
Uric acid is a heterocyclic compound of carbon, nitrogen, oxygen, and hydrogen with the formula C5H4N4O3. It forms ions and salts known as urates and acid urates, such as ammonium acid urate. Uric acid is a product of the metabolic breakdown of purine nucleotides, and it is a normal component of urine. High blood concentrations of uric acid can lead to gout and are associated with other medical conditions, including diabetes and the formation of ammonium acid urate kidney stones.

Gout is a form of inflammatory arthritis characterized by recurrent attacks of pain in a red, tender, hot, and swollen joint, caused by the deposition of needle-like crystals of uric acid known as monosodium urate crystals. Pain typically comes on rapidly, reaching maximal intensity in less than 12 hours. The joint at the base of the big toe is affected (Podagra) in about half of cases. It may also result in tophi, kidney stones, or kidney damage.
Xanthine oxidase is a form of xanthine oxidoreductase, a type of enzyme that generates reactive oxygen species. These enzymes catalyze the oxidation of hypoxanthine to xanthine and can further catalyze the oxidation of xanthine to uric acid. These enzymes play an important role in the catabolism of purines in some species, including humans.

Allopurinol is a medication used to decrease high blood uric acid levels. It is specifically used to prevent gout, prevent specific types of kidney stones and for the high uric acid levels that can occur with chemotherapy. It is taken orally or intravenously.

Phosphoglucomutase is an enzyme that transfers a phosphate group on an α-D-glucose monomer from the 1 to the 6 position in the forward direction or the 6 to the 1 position in the reverse direction.
Tumor lysis syndrome (TLS) is a group of metabolic abnormalities that can occur as a complication from the treatment of cancer, where large amounts of tumor cells are killed off (lysed) from the treatment, releasing their contents into the bloodstream. This occurs most commonly after the treatment of lymphomas and leukemias and in particular when treating non-Hodgkin lymphoma, acute myeloid leukemia, and acute lymphoblastic leukemia. This is a potentially fatal complication and people at an increased risk for TLS should be closely monitored while receiving chemotherapy and should receive preventive measures and treatments as necessary. TLS can also occur on its own although this is less common.

Lesch–Nyhan syndrome (LNS) is a rare inherited disorder caused by a deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT). This deficiency occurs due to mutations in the HPRT1 gene located on the X chromosome. LNS affects about 1 in 380,000 live births. The disorder was first recognized and clinically characterized by American medical student Michael Lesch and his mentor, pediatrician William Nyhan, at Johns Hopkins.

Glycogen storage disease type I is an inherited disease that prevents the liver from properly breaking down stored glycogen, which is necessary to maintain adequate blood sugar levels. GSD I is divided into two main types, GSD Ia and GSD Ib, which differ in cause, presentation, and treatment. There are also possibly rarer subtypes, the translocases for inorganic phosphate or glucose ; however, a recent study suggests that the biochemical assays used to differentiate GSD Ic and GSD Id from GSD Ib are not reliable, and are therefore GSD Ib.
Protein toxicity is the effect of the buildup of protein metabolic waste compounds, like urea, uric acid, ammonia, and creatinine. Protein toxicity has many causes, including urea cycle disorders, genetic mutations, excessive protein intake, and insufficient kidney function, such as chronic kidney disease and acute kidney injury. Symptoms of protein toxicity include unexplained vomiting and loss of appetite. Untreated protein toxicity can lead to serious complications such as seizures, encephalopathy, further kidney damage, and even death.
Uricosuric medications (drugs) are substances that increase the excretion of uric acid in the urine, thus reducing the concentration of uric acid in blood plasma. In general, this effect is achieved by action on the proximal tubule of the kidney. Drugs that reduce blood uric acid are not all uricosurics; blood uric acid can be reduced by other mechanisms.

Hypouricemia or hypouricaemia is a level of uric acid in blood serum that is below normal. In humans, the normal range of this blood component has a lower threshold set variously in the range of 2 mg/dL to 4 mg/dL, while the upper threshold is 530 μmol/L (6 mg/dL) for women and 619 μmol/L (7 mg/dL) for men. Hypouricemia usually is benign and sometimes is a sign of a medical condition.

Nucleic acid metabolism is a collective term that refers to the variety of chemical reactions by which nucleic acids are either synthesized or degraded. Nucleic acids are polymers made up of a variety of monomers called nucleotides. Nucleotide synthesis is an anabolic mechanism generally involving the chemical reaction of phosphate, pentose sugar, and a nitrogenous base. Degradation of nucleic acids is a catabolic reaction and the resulting parts of the nucleotides or nucleobases can be salvaged to recreate new nucleotides. Both synthesis and degradation reactions require multiple enzymes to facilitate the event. Defects or deficiencies in these enzymes can lead to a variety of diseases.

Ribose 5-phosphate (R5P) is both a product and an intermediate of the pentose phosphate pathway. The last step of the oxidative reactions in the pentose phosphate pathway is the production of ribulose 5-phosphate. Depending on the body's state, ribulose 5-phosphate can reversibly isomerize to ribose 5-phosphate. Ribulose 5-phosphate can alternatively undergo a series of isomerizations as well as transaldolations and transketolations that result in the production of other pentose phosphates as well as fructose 6-phosphate and glyceraldehyde 3-phosphate.
Purine metabolism refers to the metabolic pathways to synthesize and break down purines that are present in many organisms.
Hyperuricosuria is a medical term referring to the presence of excessive amounts of uric acid in the urine. For men this is at a rate greater than 800 mg/day, and for women, 750 mg/day. Notable direct causes of hyperuricosuria are dissolution of uric acid crystals in the kidneys or urinary bladder, and hyperuricemia. Notable indirect causes include uricosuric drugs, rapid breakdown of bodily tissues containing large quantities of DNA and RNA, and a diet high in purine.

Solute carrier family 22, member 12, also known as SLC22A12 and URAT1, is a protein which in humans is encoded by the SLC22A12 gene.
A xanthine oxidase inhibitor is any substance that inhibits the activity of xanthine oxidase, an enzyme involved in purine metabolism. In humans, inhibition of xanthine oxidase reduces the production of uric acid, and several medications that inhibit xanthine oxidase are indicated for treatment of hyperuricemia and related medical conditions including gout. Xanthine oxidase inhibitors are being investigated for management of reperfusion injury.
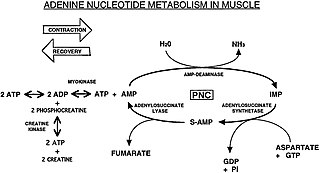
The Purine Nucleotide Cycle is a metabolic pathway in protein metabolism requiring the amino acids aspartate and glutamate. The cycle is used to regulate the levels of adenine nucleotides, in which ammonia and fumarate are generated. AMP converts into IMP and the byproduct ammonia. IMP converts to S-AMP (adenylosuccinate), which then converts to AMP and the byproduct fumarate. The fumarate goes on to produce ATP (energy) via oxidative phosphorylation as it enters the Krebs cycle and then the electron transport chain. Lowenstein first described this pathway and outlined its importance in processes including amino acid catabolism and regulation of flux through glycolysis and the Krebs cycle.

Lesinurad is a urate transporter inhibitor for treating high blood uric acid levels associated with gout. It is recommended only as an adjuvant with either allopurinol or febuxostat when these medications are not sufficient.

Gout suppressants are agents which control and prevent gout attacks after the first episode. They can be generally classified into two groups by their purpose: drugs used for induction therapy and that for maintenance therapy.