
A phosphodiesterase inhibitor is a drug that blocks one or more of the five subtypes of the enzyme phosphodiesterase (PDE), thereby preventing the inactivation of the intracellular second messengers, cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) by the respective PDE subtype(s). The ubiquitous presence of this enzyme means that non-specific inhibitors have a wide range of actions, the actions in the heart, and lungs being some of the first to find a therapeutic use.

Xanthine is a purine base found in most human body tissues and fluids, as well as in other organisms. Several stimulants are derived from xanthine, including caffeine, theophylline, and theobromine.

Adenosine (symbol A) is an organic compound that occurs widely in nature in the form of diverse derivatives. The molecule consists of an adenine attached to a ribose via a β-N9-glycosidic bond. Adenosine is one of the four nucleoside building blocks of RNA (and its derivative deoxyadenosine is a building block of DNA), which are essential for all life. Its derivatives include the energy carriers adenosine mono-, di-, and triphosphate, also known as AMP/ADP/ATP. Cyclic adenosine monophosphate (cAMP) is pervasive in signal transduction. Adenosine is used as an intravenous medication for some cardiac arrhythmias.
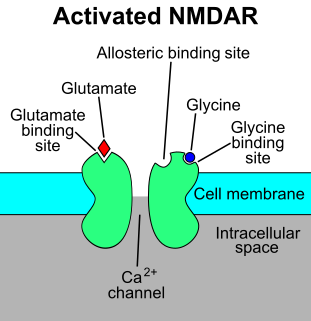
The N-methyl-D-aspartatereceptor (also known as the NMDA receptor or NMDAR), is a glutamate receptor and ion channel found in neurons. The NMDA receptor is one of three types of ionotropic glutamate receptors, the other two being AMPA and kainate receptors. Depending on its subunit composition, its ligands are glutamate and glycine (or D-serine). However, the binding of the ligands is typically not sufficient to open the channel as it may be blocked by Mg2+ ions which are only removed when the neuron is sufficiently depolarized. Thus, the channel acts as a “coincidence detector” and only once both of these conditions are met, the channel opens and it allows positively charged ions (cations) to flow through the cell membrane. The NMDA receptor is thought to be very important for controlling synaptic plasticity and mediating learning and memory functions.

The adenosine receptors (or P1 receptors) are a class of purinergic G protein-coupled receptors with adenosine as the endogenous ligand. There are four known types of adenosine receptors in humans: A1, A2A, A2B and A3; each is encoded by a different gene.
Neuroprotection refers to the relative preservation of neuronal structure and/or function. In the case of an ongoing insult the relative preservation of neuronal integrity implies a reduction in the rate of neuronal loss over time, which can be expressed as a differential equation. It is a widely explored treatment option for many central nervous system (CNS) disorders including neurodegenerative diseases, stroke, traumatic brain injury, spinal cord injury, and acute management of neurotoxin consumption. Neuroprotection aims to prevent or slow disease progression and secondary injuries by halting or at least slowing the loss of neurons. Despite differences in symptoms or injuries associated with CNS disorders, many of the mechanisms behind neurodegeneration are the same. Common mechanisms of neuronal injury include decreased delivery of oxygen and glucose to the brain, energy failure, increased levels in oxidative stress, mitochondrial dysfunction, excitotoxicity, inflammatory changes, iron accumulation, and protein aggregation. Of these mechanisms, neuroprotective treatments often target oxidative stress and excitotoxicity—both of which are highly associated with CNS disorders. Not only can oxidative stress and excitotoxicity trigger neuron cell death but when combined they have synergistic effects that cause even more degradation than on their own. Thus limiting excitotoxicity and oxidative stress is a very important aspect of neuroprotection. Common neuroprotective treatments are glutamate antagonists and antioxidants, which aim to limit excitotoxicity and oxidative stress respectively.

Levocabastine (trade name Livostin or Livocab, depending on the region) is a selective second-generation H1 receptor antagonist which was discovered at Janssen Pharmaceutica in 1979. It is used for allergic conjunctivitis.

The adenosine A1 receptor is one member of the adenosine receptor group of G protein-coupled receptors with adenosine as endogenous ligand.
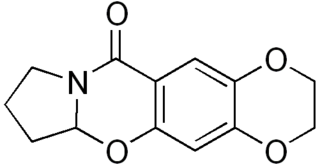
CX-614 is an ampakine drug developed by Cortex Pharmaceuticals. It has been investigated for its effect on AMPA receptors.

The adenosine A2A receptor, also known as ADORA2A, is an adenosine receptor, and also denotes the human gene encoding it.

The adenosine A2B receptor, also known as ADORA2B, is a G-protein coupled adenosine receptor, and also denotes the human adenosine A2b receptor gene which encodes it.

8-Cyclopentyl-1,3-dipropylxanthine (DPCPX, PD-116,948) is a drug which acts as a potent and selective antagonist for the adenosine A1 receptor. It has high selectivity for A1 over other adenosine receptor subtypes, but as with other xanthine derivatives DPCPX also acts as a phosphodiesterase inhibitor, and is almost as potent as rolipram at inhibiting PDE4. It has been used to study the function of the adenosine A1 receptor in animals, which has been found to be involved in several important functions such as regulation of breathing and activity in various regions of the brain, and DPCPX has also been shown to produce behavioural effects such as increasing the hallucinogen-appropriate responding produced by the 5-HT2A agonist DOI, and the dopamine release induced by MDMA, as well as having interactions with a range of anticonvulsant drugs.

ZM-241,385 is a high affinity antagonist ligand selective for the adenosine A2A receptor.
J-113,397 is an opioid drug which was the first compound found to be a highly selective antagonist for the nociceptin receptor, also known as the ORL-1 receptor. It is several hundred times selective for the ORL-1 receptor over other opioid receptors, and its effects in animals include preventing the development of tolerance to morphine, the prevention of hyperalgesia induced by intracerebroventricular administration of nociceptin, as well as the stimulation of dopamine release in the striatum, which increases the rewarding effects of cocaine, but may have clinical application in the treatment of Parkinson's disease.

SCH-442,416 is a highly selective adenosine A2a subtype receptor antagonist. It is widely used in its 11C radiolabelled form to map the distribution of A2a receptors in the brain, where they are mainly found in the striatum, nucleus accumbens, and olfactory tubercle. Given its distribution in the brain, A2a receptors have been investigated for the treatment of various neurological disorders, and SCH-442,416 has shown promise in treatment of depression, Parkinson's disease, and catalepsy.
SB-334867 is an orexin antagonist. It was the first non-peptide antagonist developed that is selective for the orexin receptor subtype OX1, with around 50x selectivity for OX1 over OX2 receptors. It has been shown to produce sedative and anorectic effects in animals, and has been useful in characterising the orexinergic regulation of brain systems involved with appetite and sleep, as well as other physiological processes. The hydrochloride salt of SB-334867 has been demonstrated to be hydrolytically unstable, both in solution and as the solid. Orexin antagonists have multiple potential clinical applications including the treatment of drug addiction, insomnia, obesity and diabetes.

PSB-10 is a drug which acts as a selective antagonist for the adenosine A3 receptor (ki value at human A3 receptor is 0.44 nM), with high selectivity over the other three adenosine receptor subtypes (ki values at human A1, A2A and A2B receptors are 4.1, 3.3 and 30 μM). Further pharmacological experiments in a [35S]GTPγS binding assay using hA3-CHO-cells indicated that PSB-10 acts as an inverse agonist (IC50 = 4 nM). It has been shown to produce antiinflammatory effects in animal studies. Simple xanthine derivatives such as caffeine and DPCPX have generally low affinity for the A3 subtype and must be extended by expanding the ring system and adding an aromatic group to give high A3 affinity and selectivity. The affinity towards adenosine A3 subtype was measured against the radioligand PSB-11.

Sonepiprazole (U-101,387, PNU-101,387-G) is a drug of the phenylpiperazine class which acts as a highly selective D4 receptor antagonist. In animals, unlike D2 receptor antagonists like haloperidol, sonepiprazole does not block the behavioral effects of amphetamine or apomorphine, does not alter spontaneous locomotor activity on its own, and lacks extrapyramidal and neuroendocrine effects. However, it does reverse the prepulse inhibition deficits induced by apomorphine, and has also been shown to enhance cortical activity and inhibit stress-induced cognitive impairment. As a result, it was investigated as an antipsychotic for the treatment of schizophrenia in a placebo-controlled clinical trial, but in contrast to its comparator olanzapine no benefits were found and it was not researched further for this indication.

CGS-15943 is a drug which acts as a potent and reasonably selective antagonist for the adenosine receptors A1 and A2A, having a Ki of 3.3nM at A2A and 21nM at A1. It was one of the first adenosine receptor antagonists discovered that is not a xanthine derivative, instead being a triazoloquinazoline. Consequently, CGS-15943 has the advantage over most xanthine derivatives that it is not a phosphodiesterase inhibitor, and so has more a specific pharmacological effects profile. It produces similar effects to caffeine in animal studies, though with higher potency.
Adenosine A2A receptor antagonists are a class of drugs that blocks adenosine at the adenosine A2A receptor. Notable adenosine A2A receptor antagonists include caffeine, theophylline and istradefylline.
