Phentermine was approved for medical use in the United States in 1959.[3] It is available as a generic medication.[3] In 2023, it was the 168th most commonly prescribed medication in the United States, with more than 3million prescriptions.[12][13] Phentermine was withdrawn from the market in the United Kingdom in 2000, while the combination medicationfen-phen, of which it was a part, was withdrawn from the market in 1997 due to side effects of fenfluramine.[14]
Medical uses
Phentermine is used for a short period of time to promote weight loss, if exercise and calorie reduction are not sufficient, and in addition to exercise and calorie reduction.[5][15]
Phentermine is approved for up to 12 weeks of use and most weight loss occurs in the first weeks.[15] However, significant loss continues through the sixth month and has been shown to continue at a slower rate through the ninth month.[16]
Tolerance usually occurs; however, risks of dependence and addiction are considered negligible.[16][18] People taking phentermine may be impaired when driving or operating machinery.[15] Consumption of alcohol with phentermine may produce adverse effects.[15]
There is currently no evidence regarding whether or not phentermine is safe for pregnant women.[5][15]
Cardiovascular effects like palpitations, tachycardia, high blood pressure, precordial pain; rare cases of stroke, angina, myocardial infarction, cardiac failure and cardiac arrest have been reported.
Central nervous system effects like overstimulation, restlessness, nervousness, insomnia, tremor, dizziness and headache; there are rare reports of euphoria followed by fatigue and depression, and very rarely, psychotic episodes and hallucinations.
Gastrointestinal effects include nausea, vomiting, dry mouth, cramps, unpleasant taste, diarrhea, and constipation.
Other adverse effects include trouble urinating, rash, impotence, changes in libido, and facial swelling.
In terms of monoamine release in vitro using rat brain synaptosomes, phentermine is about 6-fold less potent than dextroamphetamine in the case of norepinephrine release, 11-fold less potent than dextroamphetamine in the case of dopamine release, and has a ratio of norepinephrine release versus dopamine release of about 6.6:1 compared to dextroamphetamine's ratio of about 3.5:1.[8][9][10] It is more than 3-fold less potent than amphetamine in elevating brain dopamine and serotonin levels in rodents in vivo, is about 10-fold less potent than amphetamine in terms of self-administration in monkeys, and is a relatively weak reinforcer in rodents.[10][27] Although phentermine induces the release of dopamine at sufficiently high concentrations in vitro and at sufficiently high doses in rodents and monkeys in vivo, it may result in only weak or negligible brain dopamine release in humans at typical clinical doses.[9] This may be due to its selectivity for induction of norepinephrine over dopamine release and may be analogous to the case of ephedrine (which is at least 10-fold selective for induction of norepinephrine over dopamine release).[9] The effects of phentermine may be more related to noradrenergic activation rather than dopaminergic activity.[9] However, more research is needed to assess the preceding notions.[9]
As with other MRAs, phentermine produces dopaminergic neurotoxicity in rodents at high doses.[10] It can also produce serotonergic neurotoxicity at very high doses in rodents.[10] The clinical significance of these findings for humans, in which employed doses may be much lower, are unclear.[10]
The combination of phentermine with a serotonin releasing agent (SRA) like fenfluramine results in suppression of brain dopamine release by phentermine and marked attenuation or abolition of phentermine's stimulant and rewarding effects in animals and humans.[8][10][27][28][29][30][31] Conversely, combined phentermine and fenfluramine administration synergistically enhances the appetite suppression of these drugs in animals and results in greater weight loss than either drug alone in humans.[10] Fenfluramine produces serotonergic neurotoxicity in animals and addition of phentermine results in either no change or augmentation of this neurotoxicity.[10]
Phentermine has been found to be active as an agonist of the rat and human trace amine-associated receptor 1 (TAAR1).[33][34][35][36] It appears to be a weak human TAAR1 partial agonist (EC50 = 5,470nM and EmaxTooltip maximal efficacy = 68% in one study).[34] The drug shows reduced activity as a TAAR1 agonist compared to amphetamine.[34][36] TAAR1 agonism by amphetamines that possess this action may serve to auto-inhibit and constrain their effects.[37][38][39][40]
Phentermine is a very weak monoamine oxidase inhibitor (MAOI) in vitro.[41][10][42] It specifically inhibitsmonoamine oxidase A (MAO-A) (IC50Tooltip half-maximal inhibitory concentration = 85,000–143,000nM) and monoamine oxidase B (MAO-B) (IC50 = 285,000nM).[41][10][42] However, its potency as a MAOI is far below its potency as a monoamine releasing agent.[10] Relatedly, phentermine does not show neurochemical signs of MAOI activity in rodents in vivo.[10] As such, the significance of phentermine as an MAOI in humans is questionable.[10]
Pharmacokinetics
Absorption
The pharmacokinetics of phentermine are dose-dependent.[6]Peak concentrations of phentermine are reached 6hours following oral administration of a dose of 15mg.[6] The steady-state levels of phentermine with continuous administration have been found to be around 200ng/mL in clinical studies.[6] The oral bioavailability of phentermine is not affected by intake of a high-fat meal.[6]
Phentermine is a lipophilic amine that is rapidly absorbed through the gastrointestinal tract following oral ingestion.[43] Its weakly basic nature facilitates absorption in the small intestine, where it exists primarily in a non-ionized form at physiological pH. Peak plasma concentrations can vary slightly based on formulation (immediate vs. extended-release) and gastric motility.[43] The drug’s predictable absorption profile supports once-daily dosing in most therapeutic regimens.
Distribution
The volume of distribution of phentermine is 5L/kg.[6] Its plasma protein binding is approximately 17.5%.[6] This percentage of 17.5 suggests that a substantial portion of the drug remains unbound and pharmacologically active. Because of its lipophilicity and amphetamine-like structure,[43] phentermine readily crosses the blood–brain barrier, contributing to its central nervous system stimulant effects, such as increased alertness and appetite suppression. Distribution into fatty tissues may contribute to a moderate duration of action despite limited metabolism.
Metabolism
Phentermine undergoes minimal metabolism.[6] Only about 6% of an administered dose of phentermine is metabolized.[6] It is metabolized to a minor extent by para-hydroxylation, N-oxidation, and N-hydroxylation, followed by conjugation.[6]
Because phentermine’s metabolism is relatively minor, most of its pharmacologic activity is attributable to the parent compound rather than metabolites. The drug is not known to significantly induce or inhibit CYP450 enzymes,[44] suggesting a low potential for clinically meaningful drug–drug interactions. Variations in metabolic rate due to hepatic function generally have less effect on overall clearance compared to renal factors.
Elimination
The drug is eliminated mainly in urine.[6] It is excreted 62 to 85% unchanged in urine.[6] The elimination half-life of phentermine is 20 to 25hours.[6][5] The elimination of phentermine is modified by urine acidicity or pH.[6][5] In the case of acidic urine (pH < 5), the elimination half-life of phentermine has been found to be 7 to 8hours.[6] The clearance of phentermine is 8.79L/h.[6]
History
In 1959, phentermine first received approval from the US Food and Drug Administration (FDA) as an appetite suppressant.[45] Eventually a hydrochloride salt and a resin form became available.[45]
Phentermine was marketed with fenfluramine or dexfenfluramine as a combination appetite suppressant and fat burning agent under the popular name fen-phen.[46] In 1997, after 24 cases of heart valve disease in fen-phen users, fenfluramine and dexfenfluramine were voluntarily taken off the market at the request of the FDA.[47] Studies later showed nearly 30% of people taking fenfluramine or dexfenfluramine for up to 24 months had abnormal valve findings.[48]
Phentermine is still available by itself in most countries, including the US.[45] However, because it is similar to amphetamine, it is classified as a controlled substance in many countries. Internationally, phentermine is a schedule IV drug under the Convention on Psychotropic Substances.[49] In the United States, it is classified as a Schedule IV controlled substance under the Controlled Substances Act. In contrast, amphetamine preparations are classified as Schedule II controlled substances.[50]
A company called Vivus developed a combination drug, phentermine/topiramate that it originally called Qnexa and then called Qsymia, which was invented and used off-label by Thomas Najarian, who opened a weight-clinic in Los Osos, California in 2001; Najarian had previously worked at Interneuron Pharmaceuticals, which had developed one of the fen-phen drugs previously withdrawn from the market.[51] The FDA rejected the combination drug in 2010 due to concerns over its safety.[51] In 2012 the FDA approved it after Vivus re-applied with further safety data.[52] At the time, one obesity specialist estimated that around 70% of his colleagues were already prescribing the combination off-label.[51]
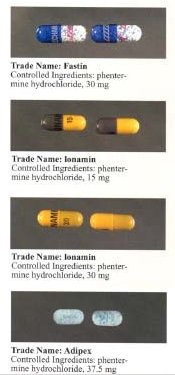
Phentermine is marketed under many brand names and formulations worldwide, including Acxion, Adipex, Adipex-P, Duromine, Elvenir, Fastin, Ionamin, Lomaira (phentermine hydrochloride), Panbesy, Qsymia (phentermine and topiramate), Razin, Redusa, Sentis, Suprenza, and Terfamex.[56]
12Partilla JS, Dersch CM, Baumann MH, Carroll FI, Rothman RB (1999). "Profiling CNS Stimulants with a High-Throughput Assay for Biogenic Amine Transporter Substrates". Problems of Drug Dependence 1999: Proceedings of the 61st Annual Scientific Meeting, The College on Problems of Drug Dependence, Inc(PDF). NIDA Res Monogr. Vol.180. pp.1–476 (252). PMID11680410. Archived from the original(PDF) on 5 August 2023. RESULTS. Methamphetamine and amphetamine potently released NE (IC50s = 14.3 and 7.0 nM) and DA (IC50s = 40.4 nM and 24.8 nM), and were much less potent releasers of 5-HT (IC50s = 740 nM and 1765 nM). Phentermine released all three biogenic amines with an order of potency NE (IC50 = 28.8 nM)> DA (IC50 = 262 nM)> 5-HT (IC50 = 2575 nM). [...] Chlorphentermine was a very potent 5-HT releaser (IC50 = 18.2 nM), a weaker DA releaser (IC50 = 935 nM) and inactive in the NE release assay. Chlorphentermine was a moderate potency inhibitor of [3H]NE uptake (Ki = 451 nM). [...]
↑Partilla JS, Dempsey AG, Nagpal AS, Blough BE, Baumann MH, Rothman RB (October 2006). "Interaction of amphetamines and related compounds at the vesicular monoamine transporter". J Pharmacol Exp Ther. 319 (1): 237–246. doi:10.1124/jpet.106.103622. PMID16835371.
↑Brauer LH, Johanson CE, Schuster CR, Rothman RB, de Wit H (April 1996). "Evaluation of phentermine and fenfluramine, alone and in combination, in normal, healthy volunteers". Neuropsychopharmacology. 14 (4): 233–241. doi:10.1016/0893-133X(95)00113-R. PMID8924191.
12Bunzow JR, Sonders MS, Arttamangkul S, Harrison LM, Zhang G, Quigley DI, etal. (December 2001). "Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor". Mol Pharmacol. 60 (6): 1181–1188. doi:10.1124/mol.60.6.1181. PMID11723224.
↑Espinoza S, Gainetdinov RR (2014). "Neuronal Functions and Emerging Pharmacology of TAAR1". Taste and Smell. Topics in Medicinal Chemistry. Vol.23. Cham: Springer International Publishing. pp.175–194. doi:10.1007/7355_2014_78. ISBN978-3-319-48925-4. Interestingly, the concentrations of amphetamine found to be necessary to activate TAAR1 are in line with what was found in drug abusers [3, 51, 52]. Thus, it is likely that some of the effects produced by amphetamines could be mediated by TAAR1. Indeed, in a study in mice, MDMA effects were found to be mediated in part by TAAR1, in a sense that MDMA auto-inhibits its neurochemical and functional actions [46]. Based on this and other studies (see other section), it has been suggested that TAAR1 could play a role in reward mechanisms and that amphetamine activity on TAAR1 counteracts their known behavioral and neurochemical effects mediated via dopamine neurotransmission.
↑Kuropka P, Zawadzki M, Szpot P (May 2023). "A narrative review of the neuropharmacology of synthetic cathinones-Popular alternatives to classical drugs of abuse". Hum Psychopharmacol. 38 (3) e2866. doi:10.1002/hup.2866. PMID36866677. Another feature that distinguishes [synthetic cathinones (SCs)] from amphetamines is their negligible interaction with the trace amine associated receptor 1 (TAAR1). Activation of this receptor reduces the activity of dopaminergic neurones, thereby reducing psychostimulatory effects and addictive potential (Miller, 2011; Simmler et al., 2016). Amphetamines are potent agonists of this receptor, making them likely to self‐inhibit their stimulating effects. In contrast, SCs show negligible activity towards TAAR1 (Kolaczynska et al., 2021; Rickli et al., 2015; Simmler et al., 2014, 2016). [...] It is worth noting, however, that for TAAR1 there is considerable species variability in its interaction with ligands, and it is possible that the in vitro activity of [rodent TAAR1 agonists] may not translate into activity in the human body (Simmler et al., 2016). The lack of self‐regulation by TAAR1 may partly explain the higher addictive potential of SCs compared to amphetamines (Miller, 2011; Simmler et al., 2013).
↑Simmler LD, Buser TA, Donzelli M, Schramm Y, Dieu LH, Huwyler J, etal. (January 2013). "Pharmacological characterization of designer cathinones in vitro". Br J Pharmacol. 168 (2): 458–470. doi:10.1111/j.1476-5381.2012.02145.x. PMC3572571. PMID22897747. β-Keto-analogue cathinones also exhibited approximately 10-fold lower affinity for the TA1 receptor compared with their respective non-β-keto amphetamines. [...] Activation of TA1 receptors negatively modulates dopaminergic neurotransmission. Importantly, methamphetamine decreased DAT surface expression via a TA1 receptor-mediated mechanism and thereby reduced the presence of its own pharmacological target (Xie and Miller, 2009). MDMA and amphetamine have been shown to produce enhanced DA and 5-HT release and locomotor activity in TA1 receptor knockout mice compared with wild-type mice (Lindemann et al., 2008; Di Cara et al., 2011). Because methamphetamine and MDMA auto-inhibit their neurochemical and functional effects via TA1 receptors, low affinity for these receptors may result in stronger effects on monoamine systems by cathinones compared with the classic amphetamines.
Notes: (1) TAAR1 activity of ligands varies significantly between species. Some agents that are TAAR1 ligands in some species are not in other species. This navbox includes all TAAR1 ligands regardless of species. (2) See the individual pages for references, as well as the List of trace amines, TAAR, and TAAR1 pages. See also:Receptor/signaling modulators
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.