Mechanism of action
This section may be too technical for most readers to understand.(January 2016) |

The 5-HT2C receptors are G protein–coupled receptors that are coupled to phospholipase C (PLC) via Gαq, phospholipase A2 (PLA2), and possibly Gα13. PLC metabolizes phosphatidylinositol 4,5-bisphosphate into inositol 1,4,5-triphosphate (IP3). IP3 regulates cellular Ca2+ flux by binding to IP3 receptors, inducing Ca2+ release. In addition, the activation of PLA2 also results in recruitment of a RhoA/PLD pathway through RhoA, an enzyme that regulates a wide spectrum of cellular functions through PLD (phospholipase D) target protein. The 5-HT2C receptors can also stimulate the extracellular signal-regulated kinase (ERK) pathway which is activated by neurotrophins and other neuroactive chemicals. Production of these chemicals effects neuronal differentiation, survival, regeneration, and structural and functional plasticity. Early studies of the ERK pathway showed that mood stabilizers for the treatment of manic-depressive illness stimulated the pathway. This led to the understanding that stimulation of the 5-HT2C receptors could regulate manic-depressive conditions in a manner similar to mood stabilizers. [23] [24] [25] [3]
5-HT2C receptors are located only within the CNS, where they can be found in several locations. The highest density of receptor expression is within the choroid plexus. Other brain locations include the nucleus of the solitary tract, dorsomedial hypothalamus, paraventricular hypothalamic nucleus and the amygdala, all of which are associated with regulation of food intake. This distribution pattern may explain the effect they have in integral function in the control of many physiological and behavioral responses, such as feeding, anxiety, temperature regulation, locomotion, sexual behavior, and the occurrence of seizures. [26] [27]
Binding
The 5-HT2C receptors and ligand binding

5-HT2 receptors are G protein-coupled receptors that can regulate cellular signaling in the absence of a ligand. This can be explained by a two-state model (Figure 2) where the receptor is in equilibrium between two states, an active state (R*) and an inactive state (R). Basal effector activity is defined, in part, by the absolute level of (R*), which will increase along with increasing receptor density. Ligands that preferentially bind to and stabilize the R state are termed inverse agonists and reduce the effector activity. Agonists preferentially bind to and stabilize the R* state, thereby increasing effector activity. Neutral antagonists show equal affinity for both conformations and do not alter the equilibrium between the two states, however they occupy the receptor and can block the effect of both agonists and inverse agonists. [28] [29]
5-HT2C and 5-HT2A receptors have a similar amino acid sequence homology, with ~50% overall sequence identity and ~80% within the TM domains, resulting in a similar pharmacological profile for the two receptors. Both receptors couple the same cellular signal transduction pathways, PLC and PLA2, that lead to an accumulation of inositol phosphate and Ca2+ within the postsynaptic cell. [28]
The 5-HT2C receptors are the only G-protein coupled receptors known to undergo a post-transcriptional process of RNA editing. The 5-HT2C receptor gene is found on the X-chromosome, Xq24. This gene product undergoes an RNA editing process leading to a decrease in agonist binding affinity, however antagonist binding remains unaltered. This process of RNA editing generates 14 unique receptor isoforms of the 5-HT2C receptor that differ in three amino acids in the second intracellular loop. [28] [30]
Serotonin binding to 5-HT2C
Serotonin is an endogenous non-selective agonist for the 5-HT2C receptor with a binding constant of Ki = 16.0 nM. When serotonin binds to the receptors, the most important contacts are in TM helixes 3, 5 and 6 (Figure 3), while the other four TM helixes do not interact directly with the serotonin compound. When binding of serotonin takes place, the protonated primary amine site forms a salt bridge with D134 residue in TM 3, as well as forming a hydrogen bond with residue S138 in TM 3. The aromatic indole ring forms a strong Van der Waals interaction with residues F223 in TM 5 and F328 in TM 6. The ring falls tight into the receptor pocket, stacked between two phenylalanines. Amine of the indole group forms a hydrogen bond with S219 residue in TM 5 and hydroxide substituent of the indole forms hydrogen bonds both with residue S131 in TM 3 and I332 in TM 6. There is also a strong Van der Waals interaction between the indole and I332 in TM 6. [31]

Pharmacophore
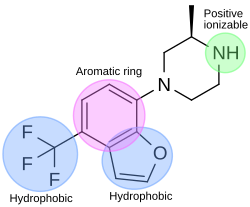
In the drug discovery process of a 5-HT2C agonist, a pharmacophore module has been used to discover novel 5-HT2C receptor ligands. The pharmacophore has four features; one aromatic ring, two hydrophobic features and one positive ionizable feature. Figure 4 shows an example of a compound that fits the agonist pharmacophore perfectly. The nitrogen atom of piperazine fits the positive ionizable feature, the benzofuran part fits the aromatic ring and one hydrophobic, and the trifluoromethane part fits another hydrophobic feature of the pharmacophore. [32]
Structure-activity relationships
In a virtual screen for novel agonists, a structure-activity relationship was determined from the most potent compounds ('hits') identified.[ clarification needed ] These hits contained a pyrazolo[3,4-d]pyrimidine core (shown in figure 5), which is important for potency toward the 5-HT2C receptors. Compounds with maximum potency featured two substituents linked to the core structure. The first substituent is a piperazine ring, containing a small hydrophobic group; the second substituent is a phenyl part containing a halogen- and/or oxygen-containing side chain (electronegative groups), see derivatives 1 and 2 in figure 5. Addition of aromatic groups to the piperazine ring reduces potency (derivative 4 in figure 5) and the absence of the piperazine ring or substitution with other aliphatic- or cyclic groups reduces potency as well (derivatives 5 and 6 in figure 5). [32]









A series of 3-benzazepine derivatives, such as Lorcaserin (Figure 6) have been evaluated for their potency and selectivity for the 5-HT2C receptors. Lorcaserin is a very potent agonist, but the potency is dependent on the presence of a chloro substituent in position 8. [7] [33] [34]
Arylpiperazine-containing compounds such as mCPP (Figure 7), show good potency toward the 5-HT2C receptors, but do not have sufficient selectivity for the 5-HT2C receptors over the other two receptor subtypes. Many derivatives have been examined in an attempt to increase the selectivity. Derivatives lacking the arylpiperazine core, such as 4-aryl-1,2,3,6-tetrahydropyridinum chlorine analogues, are more favorable for potency and selectivity over the other two receptors (Figure 7). [35]
Functional selectivity
In 2016 the discovery of novel G protein biased 5-HT2C receptor agonists was published. [36]