In biology and biochemistry, the active site is the region of an enzyme where substrate molecules bind and undergo a chemical reaction. The active site consists of amino acid residues that form temporary bonds with the substrate, the binding site, and residues that catalyse a reaction of that substrate, the catalytic site. Although the active site occupies only ~10–20% of the volume of an enzyme, it is the most important part as it directly catalyzes the chemical reaction. It usually consists of three to four amino acids, while other amino acids within the protein are required to maintain the tertiary structure of the enzymes.
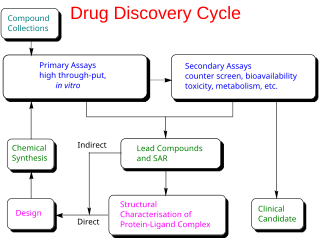
Drug design, often referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques. This type of modeling is sometimes referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design. In addition to small molecules, biopharmaceuticals including peptides and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also been developed.
Quantitative structure–activity relationship models are regression or classification models used in the chemical and biological sciences and engineering. Like other regression models, QSAR regression models relate a set of "predictor" variables (X) to the potency of the response variable (Y), while classification QSAR models relate the predictor variables to a categorical value of the response variable.

Medicinal or pharmaceutical chemistry is a scientific discipline at the intersection of chemistry and pharmacy involved with designing and developing pharmaceutical drugs. Medicinal chemistry involves the identification, synthesis and development of new chemical entities suitable for therapeutic use. It also includes the study of existing drugs, their biological properties, and their quantitative structure-activity relationships (QSAR).

Selective estrogen receptor modulators (SERMs), also known as estrogen receptor agonist/antagonists (ERAAs), are a class of drugs that act on the estrogen receptor (ER). A characteristic that distinguishes these substances from pure ER agonists and antagonists is that their action is different in various tissues, thereby granting the possibility to selectively inhibit or stimulate estrogen-like action in various tissues.

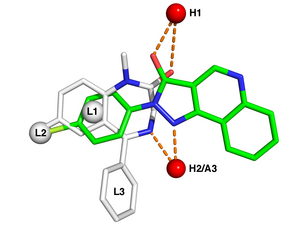
In biochemistry and pharmacology, a ligand is a substance that forms a complex with a biomolecule to serve a biological purpose. The etymology stems from Latin ligare, which means 'to bind'. In protein-ligand binding, the ligand is usually a molecule which produces a signal by binding to a site on a target protein. The binding typically results in a change of conformational isomerism (conformation) of the target protein. In DNA-ligand binding studies, the ligand can be a small molecule, ion, or protein which binds to the DNA double helix. The relationship between ligand and binding partner is a function of charge, hydrophobicity, and molecular structure.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.

Macrocycles are often described as molecules and ions containing a ring of twelve or more atoms. Classical examples include the crown ethers, calixarenes, porphyrins, and cyclodextrins. Macrocycles describe a large, mature area of chemistry.
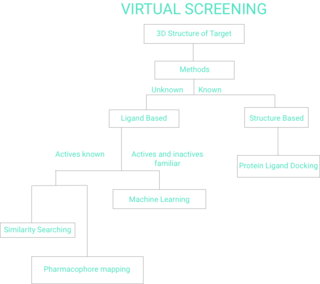
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.
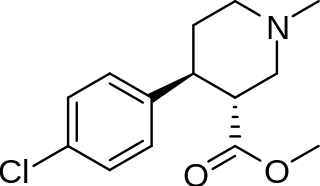
(+)-CPCA is a stimulant drug similar in structure to pethidine and to RTI-31, but nocaine is lacking the two-carbon bridge of RTI-31's tropane skeleton. This compound was first developed as a substitute agent for cocaine.
In the fields of computational chemistry and molecular modelling, scoring functions are mathematical functions used to approximately predict the binding affinity between two molecules after they have been docked. Most commonly one of the molecules is a small organic compound such as a drug and the second is the drug's biological target such as a protein receptor. Scoring functions have also been developed to predict the strength of intermolecular interactions between two proteins or between protein and DNA.

Chemical Computing Group is a software company specializing in research software for computational chemistry, bioinformatics, cheminformatics, docking, pharmacophore searching and molecular simulation. The company's main customer base consists of pharmaceutical and biotechnology companies, as well as academic research groups. It is a private company that was founded in 1994; it is based in Montreal, Quebec, Canada. Its main product, Molecular Operating Environment (MOE), is written in a self-contained programming system, the Scientific Vector Language (SVL).
LigandScout is computer software that allows creating three-dimensional (3D) pharmacophore models from structural data of macromolecule–ligand complexes, or from training and test sets of organic molecules. It incorporates a complete definition of 3D chemical features that describe the interaction of a bound small organic molecule (ligand) and the surrounding binding site of the macromolecule. These pharmacophores can be overlaid and superimposed using a pattern-matching based alignment algorithm that is solely based on pharmacophoric feature points instead of chemical structure. From such an overlay, shared features can be interpolated to create a so-called shared-feature pharmacophore that shares all common interactions of several binding sites/ligands or extended to create a so-called merged-feature pharmacophore. The software has been successfully used to predict new lead structures in drug design, e.g., predicting biological activity of novel human immunodeficiency virus (HIV) reverse transcriptase inhibitors.
A cannabinoid receptor antagonist, also known simply as a cannabinoid antagonist or as an anticannabinoid, is a type of cannabinoidergic drug that binds to cannabinoid receptors (CBR) and prevents their activation by endocannabinoids. They include antagonists, inverse agonists, and antibodies of CBRs. The discovery of the endocannabinoid system led to the development of CB1 receptor antagonists. The first CBR inverse agonist, rimonabant, was described in 1994. Rimonabant blocks the CB1 receptor selectively and has been shown to decrease food intake and regulate body-weight gain. The prevalence of obesity worldwide is increasing dramatically and has a great impact on public health. The lack of efficient and well-tolerated drugs to cure obesity has led to an increased interest in research and development of CBR antagonists. Cannabidiol (CBD), a naturally occurring cannabinoid and a non-competitive CB1/CB2 receptor antagonist, as well as Δ9-tetrahydrocannabivarin (THCV), a naturally occurring cannabinoid, modulate the effects of THC via direct blockade of cannabinoid CB1 receptors, thus behaving like first-generation CB1 receptor inverse agonists, such as rimonabant. CBD is a very low-affinity CB1 ligand, that can nevertheless affect CB1 receptor activity in vivo in an indirect manner, while THCV is a high-affinity CB1 receptor ligand and potent antagonist in vitro and yet only occasionally produces effects in vivo resulting from CB1 receptor antagonism. THCV has also high affinity for CB2 receptors and signals as a partial agonist, differing from both CBD and rimonabant.
Triptans is a word commonly used for a class of anti-migraine drugs that are selective 5-hydroxytryptamine/serotonin1B/1D (5-HT1B/1D) agonists. Migraine is a complex disease which affects about 15% of the population and can be highly disabling. Triptans have advantages over ergotamine and dihydroergotamine, such as selective pharmacology, well established safety record and evidence-based prescribing instructions. Triptans are therefore often preferred treatment in migraine.
CCR5 receptor antagonists are a class of small molecules that antagonize the CCR5 receptor. The C-C motif chemokine receptor CCR5 is involved in the process by which HIV, the virus that causes AIDS, enters cells. Hence antagonists of this receptor are entry inhibitors and have potential therapeutic applications in the treatment of HIV infections.
Matched molecular pair analysis (MMPA) is a method in cheminformatics that compares the properties of two molecules that differ only by a single chemical transformation, such as the substitution of a hydrogen atom by a chlorine one. Such pairs of compounds are known as matched molecular pairs (MMP). Because the structural difference between the two molecules is small, any experimentally observed change in a physical or biological property between the matched molecular pair can more easily be interpreted. The term was first coined by Kenny and Sadowski in the book Chemoinformatics in Drug Discovery.
Chemoproteomics entails a broad array of techniques used to identify and interrogate protein-small molecule interactions. Chemoproteomics complements phenotypic drug discovery, a paradigm that aims to discover lead compounds on the basis of alleviating a disease phenotype, as opposed to target-based drug discovery, in which lead compounds are designed to interact with predetermined disease-driving biological targets. As phenotypic drug discovery assays do not provide confirmation of a compound's mechanism of action, chemoproteomics provides valuable follow-up strategies to narrow down potential targets and eventually validate a molecule's mechanism of action. Chemoproteomics also attempts to address the inherent challenge of drug promiscuity in small molecule drug discovery by analyzing protein-small molecule interactions on a proteome-wide scale. A major goal of chemoproteomics is to characterize the interactome of drug candidates to gain insight into mechanisms of off-target toxicity and polypharmacology.
Yvonne Connolly Martin is an American cheminformatics and computer-aided drug design expert who rose to the rank of Senior Volwiler Research Fellow at Abbott Laboratories. Trained in chemistry at Northwestern University, she became a leader in collaborative science aimed at discovering and developing bioactive molecules as therapeutic agents, with her contributions proceeding from application of methods to understand how descriptors of molecular shapes and physicochemical properties relate to their biological activity. She is the author of a seminal volume in cheminformatics, Quantitative Drug Design, and has been the recipient of numerous awards in her field, including being named as a fellow of the American Association for the Advancement of Science (1985) and of the International Union of Pure and Applied Chemistry (2000), and receiving the Herman Skolnik Award (2009) and the Award for Computers in Chemical and Pharmaceutical Research (2017) from the American Chemical Society.
Molecular Operating Environment (MOE) is a drug discovery software platform that integrates visualization, modeling and simulations, as well as methodology development, in one package. MOE scientific applications are used by biologists, medicinal chemists and computational chemists in pharmaceutical, biotechnology and academic research. MOE runs on Windows, Linux, Unix, and macOS. Main application areas in MOE include structure-based design, fragment-based design, ligand-based design, pharmacophore discovery, medicinal chemistry applications, biologics applications, structural biology and bioinformatics, protein and antibody modeling, molecular modeling and simulations, virtual screening, cheminformatics & QSAR. The Scientific Vector Language (SVL) is the built-in command, scripting and application development language of MOE.