Discovery and development of phosphodiesterase 5 inhibitors
Last updated
Drug discovery
This article needs to be updated. Please help update this article to reflect recent events or newly available information.(March 2017)
Phosphodiesterases (PDEs) are a superfamily of enzymes. This superfamily is further classified into 11 families, PDE1 - PDE11, on the basis of regulatory properties, amino acid sequences, substrate specificities, pharmacological properties and tissue distribution. Their function is to degrade intracellular second messengers such as cyclic adenine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) which leads to several biological processes like effect on intracellular calcium level by the Ca2+ pathway.[1]
Phosphodiesterase 5 (PDE5) is widely expressed in several tissues in the body for example brain, lung, kidney, urinary bladder, smooth muscle and platelets.[1] It is possible to prevent cGMP hydrolysis by inhibiting PDE5 and therefore treat diseases associated with low cGMP levels; because of this, PDE5 is an ideal target for the development of inhibitors.[2] The therapeutic effects of PDE5 inhibition have been demonstrated in several cardiovascular conditions, chronic kidney disease and diabetes mellitus.[3]
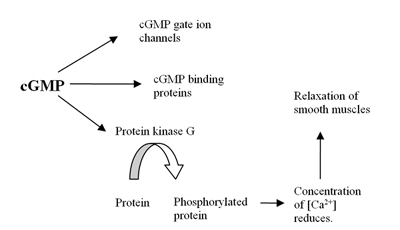
The human genome contains at least 21 genes involved in determining the intracellular levels of cAMP and cGMP by the expression of phosphodiesteraseproteins or PDE's. These PDE's are grouped into at least 11 functional subfamilies, named PDE1-PDE11.[4] PDEs are enzymes that hydrolyzecyclic adenosine 3,5-monophosphate (cAMP) and cyclic guanosine 3,5-monophospahate (cGMP), which are intracellular second messengers, into AMP and GMP. These second messengers control many physiological processes.[5] The cAMP is formed from ATP by the enzymeadenylyl cyclase and cGMP is formed from GTP by the enzyme guanylyl cyclase which are either membrane bound or soluble in the cytosol. When soluble it functions as a receptor for nitric oxide (NO) (see figure 1).[6] Formation of cGMP initiates several reactions in the body including influence on cGMP ion channels, cGMP binding proteins and protein kinase G (PKG). The effect on PKG reduces levels of calcium leading to relaxation of smooth muscles (see figure 2).[7] The PDE5 enzyme is specific for cGMP which means it only hydrolyzes cGMP but not cAMP.[8] The selectivity is mediated through an intricate network of hydrogen bonding which is favorable for cGMP but unfavorable for cAMP in PDE5.[9] By inhibition of PDE5 enzyme the cGMP concentration will be raised and can therefore increase the relaxation of smooth muscles.[7] PDE5 has only one subtype, PDE5A, of which there are 4 isoforms in humans called PDE5A1-4.[8] The difference in PDE5A1-3 isoforms is only in the 5´ end of the mRNA and corresponding N-terminal of the protein.[10]
Figure 1: Effect of PDE5 enzymeFigure 2. Formation of cGMP and its effect in the body
PDE enzymes are composed of 3 functional domains: an N-terminal cyclin fold domain, a linker helical domain and a C-terminal helical bundle domain (see figure 3).[9] The active site is a deep pocket at the junction of the 3 subdomains and is lined with highly conserved residues between isotypes of PDE.[14] The pocket is approximately 15 Å deep and the opening is approximately 20 by 10 Å. The volume of the active site has been calculated to be between 875 and 927 Å3.[14] The active site of PDE5 has been described as subdivided into 3 main regions based on its crystal structure in complex with sildenafil:[7]
M site: contains both a zinc and magnesium ion. The role of the ions is to stabilize the structure and activation of hydroxide to mediate the reaction. Current PDE5 inhibitors do not interact with the metal ions, in contrast with cGMP. Direct or indirect interactions may improve the potency of future inhibitors.[9]
Q pocket: it is believed that the guanidine group of cGMP binds in this region as the Q pocket accommodates the pyrazolopyrimidinone group (see figure 4) of sildenafil. The pyrazolopyrimidinone of sildenafil mimics that of the guanine in cGMP and has the same H-bond donor and acceptor features, forming a bidendrate H-bond with Q817. Card et al. describe the Q pocket as subdivided into 3 parts:[14]
A saddle formed by the conserved glutamine (Q817 in PDE5A, Q443 in PDE4B and Q369 in PDE4D) and the P clamp (a hydrophobic clamp at the narrow side of the active sites pocket, formed of invariant purine-selective glutamine and a pair of conserved residues).[14]
2 narrow, hydrophobic pockets, Q1 and Q2, composed mainly of hydrophobic residues flanking the saddle.[14]
L region: the methyl piperazine group (see figure 4) of sildenafil is surrounded by Tyr 664, Met 816, Ala 823 and Gly 819 residues, and residues 662-664 form a lid over the pocket narrowing the entrance to the active site of PDE5.
Jeon et al.[9] also describe a fourth pocket called the H pocket which is hydrophobic and accommodates the ethoxy phenyl group of sildenafil The 3 PDE5 inhibitors already on the market, sildenafil, tadalafil and vardenafil, occupy part of the active site, mainly around the Q pocket and sometimes the M pocket as well and all 3 interact with the active site in 3 important manners:
interaction between the metal ions mediated through water
hydrophobic interaction with hydrophobic residues lining the cavity of the active site.[14]
It has also been described that the hydrophobic interaction with the Q1 and Q2 pockets are important for inhibitor potency and differences between isotypes of PDE in the Q2 pocket can be exploited for selectivity between isotypes.[14]
Figure 3: Vardenafil (yellow) is seen here in complex with PDE5. Violet: N-terminal domain, grey: linker domain, blue: C-terminal domain, magnesium and zinc ions can also be seen. PDB refcode 1XP0. Image produced by Cn3D12 from (https://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?form=6&db=t&Dopt=s&uid=31153)Figure 4: Sildenafil. Blue shows the methyl piperazine group, orange is the ethoxyphenyl group and orange and green together represent the pyrazolopyrimidinone group.
Role in diseases
Erectile dysfunction
Drugs that inhibit PDE5, sildenafil, tadalafil and vardenafil, have been used as treatment for erectile dysfunction.[16] These inhibitors increase the cGMP, smooth muscle relaxation and consequently cause penis erection[9] during sexual stimulation.[17]
Pulmonary arterial hypertension
Upregulation of PDE5 gene expression has been observed in animal models of pulmonary hypertension, and is thought to contribute to vasoconstriction in the lung.[3] Several randomised controlled trials investigating PDE5 inhibitors use in pulmonary arterial hypertension, a subtype of pulmonary hypertension, have demonstrated their potent effects in reducing pulmonary hypertension and vascular remodelling and improving symptoms and mortality in patients with the condition.[3][7][18] Long-term treatment with a PDE5 inhibitor has been shown to enhance natriuretic peptide-cGMP pathway, downregulate Ca2+ signaling pathway and alter vascular tone in pulmonary arteries in rat models.[9]
Benign prostatic hyperplasia
As of 2011, the long-acting agent tadalafil is licensed for the treatment of urinary symptoms resulting from benign prostatic hyperplasia.[3]
Future indications for PDE5 inhibitors
Cardiovascular diseases
PDE5 inhibitors have broad-ranging effects on the cardiovascular system beyond their acute haemodynamic influence. For example, PDE5 inhibitors have been shown to improve several parameters of endothelial function.[3] Increasingly, their use in the management of systemic hypertension (including treatment-resistant hypertension), cardioprotection, heart failure, and peripheral arterial disease are being evaluated.[3]
Heart failure
PDE5 inhibitors have shown promise in the treatment of heart failure with reduced ejection fraction through several beneficial effects on lung vasculature, cardiac remodelling and diastolic function.[3] A study showed that effective treatment of pulmonary arterial hypertension with sildenafil improved functional capacity and reduced right ventricular mass in patients. The effects on right ventricular remodeling were significantly greater in comparison with the non-selective endothelial receptor antagonist bosentan.[7] However, PDE5 inhibitors may be harmful in patients with heart failure with preserved ejection fraction due to potential negative inotropic effects.[3]
Chronic kidney disease
Experimental studies in animals have shown that PDE5 inhibitors may reverse kidney damage independently of their effects on blood pressure through intra-renal mechanisms.[3] In humans, PDE5 inhibitors have also been shown to reduce proteinuria, a marker of kidney damage.[3] However, the successful introduction of SGLT2 inhibitors and endothelin receptor antagonists to the field of renal therapeutics makes the development of PDE5 inhibitors for this purpose unlikely.[3]
Diabetes mellitus
PDE5 inhibitors have been shown to have various macrovascular, microvascular and metabolic benefits in diabetes mellitus,[3] and in a large study of men with type 2 diabetes mellitus the agents were found to significantly reduce patients' risk of death from any cause.[19] It is unclear to what extent this observation reflects the protective effects of PDE5 inhibitors against cardiovascular and renal disease.[3]
Raynaud's phenomenon
Sildenafil has been shown to be at least as effective as calcium channel blockers in treating severe Raynaud's phenomenon (RP) associated with systemic sclerosis and digital ulceration.[3] When given sildenafil for 4 weeks subjects had reduced mean frequency and duration of Raynaud attacks and a significantly lowered mean Raynaud's condition score. The capillary blood flow velocity also increased in each individual patient and the mean capillary flow velocity of all patients increased significantly. These results came without significant reductions of the systemic blood pressure.[7] However, the therapeutic effects of PDE5 inhibitors in primary (idiopathic) RP are less well defined.[3]
Stroke
Sildenafil has been shown to significantly improve neurovascular coupling without affecting overall cerebral blood flow by increasing brain levels of cGMP, evoking neurogenesis and reducing neurological deficits in rats 2 or 24 hours after stroke. These experimental data suggest that PDE5 inhibitors may have a role in promoting recovery from stroke.[7][9][11] However, studies in humans remain inconclusive.[3]
Premature ejaculation
Adding PDE5 inhibitors to SSRI drugs (e.g. paroxetine) for the treatment of premature ejaculation could result in better ejaculatory control according to recent studies.[11] Possible mechanism is based on nitric oxide (NO)/cGMP transduction system as a central and peripheral mediator of inhibitory non-adrenergic, non-cholinergic nitrergic neurotransmission in the urogenital system.[16]
Female sexual arousal disorder
PDE5 is expressed in clitoral corpus cavernosum and in vaginal smooth muscle and epithelium. Therefore, it is possible that PDE5 inhibitors could affect female sexual arousal disorder but further research is needed. Increased levels of cGMP have been shown to occur in human-cultured vaginal smooth muscle cells treated with a PDE5 inhibitor suggesting involvement of the NO/cGMP axis in the female sexual response.[11]
Sexual Exhaustion Disorder
The similarity of many PDE5 inhibitors to the structure of many of the analogs of caffeine that are also adenosine antagonists suggests that in the future, it may be possible to design an PDE5 inhibitor that, like caffeine, is also an adenosine antagonist.
Discovery
PDE5 is an enzyme that was first purified in 1980 from a rats lung.[20] PDE5 converts intracellular cGMP to the nucleotide GMP.[21] Many tissues contain PDE5, such as lungs, kidneys, brain, platelets, liver, prostate, urethra, bladder and smooth muscles. Because of the localization of PDE5 in the smooth muscle tissue, inhibitors were developed for the treatment of erectile dysfunction along with pulmonary hypertension.[1][2]
Sildenafil was initially introduced for clinical trial in 1989. It was the result of extensive research on chemical agents targeting PDE5 that could be effective in treatment of coronary heart disease.[22] Sildenafil did not prove effective for coronary heart disease but an interesting side effect was discovered, a penile erection. That side effect soon became the main field of investigation.[23] The inhibitor is highly selective for the PDE5 family.[22]
Sildenafil is a prototype of PDE5 inhibitors that Pfizer launched as Viagra. It was approved by the Food and Drug Administration (FDA) in 1998 as the first oral medicine for erectile dysfunction. Later, in the year 2005, it was approved for the treatment of pulmonary arterial hypertension.[2] Vardenafil and tadalafil were discovered in 1990. These drugs came out of research programs focusing on finding PDE5 inhibitors for the treatment of cardiovascular diseases and erectile dysfunction. The two PDE5 inhibitors soon became treatments for these conditions.[22][23]
Tadalafil is the most versatile inhibitor and has the longest half-life, 17.5 hours. This allows for a longer therapeutic window and is therefore often a more convenient drug than others with a shorter therapeutic window. Tadalafil is more bioavailable (80%) than sildenafil (40%) and vardenafil (15%) but it has a slow absorption, or about 2 hours compared to 50 minutes of sildenfil. Vardenafil is most known for its potency.[24]
Because of severe adverse effects and patients dissatisfaction with current therapy choices other inhibitors have recently been approved for clinical use. These inhibitors are udenfil, avanafil lodenafil and mirodenafil.[25]
Development
Biological activity
Penile erection
Figure 1. Biological pathway of penile erection
Penile erection is a hemodynamic event in the smooth muscle of corpus cavernous.[26] PDE5 is the main cGMP hydrolysing enzyme found in penile corpus cavernous.[27] Erection is triggered by release of the neurotransmitternitric oxide (NO) from non-adrenergic and non-cholinergic neurons from nerve ending in the penis as well as from endothelial cells. NO activates soluble guanylyl cyclase in smooth muscle cells in the penis which results in increased production of 3'-5'-cyclic guanosine monophosphate from guanosine-5'-triphosphate (GTP).[21][28][29] Cyclic GMP binds to the cGMP-dependent protein kinase (PKG1) which phosphorylates several proteins that results in decreased intracellular calcium. Lower intracellular calcium leads to smooth muscle relaxation and ultimately penile erection. This pathway is demonstrated in figure 1.[29][30]
Erectile dysfunction
PDE5 degrades cGMP and therefore inhibits erection. As demonstrated in figure 1, inhibition of PDE5 reduces degradation of cGMP and leads to penile erection.[28][31] Because of this action PDE5 inhibitors have been developed for the treatment of penile erectile dysfunction.[32]
The phosphodiesterase 5 enzyme
Figure 2. PDE5-Inhibitors interfere with the conversion of cGMP into 5'-GMP which then has downstream effects in the cell.
The PDE5 enzyme has a molecular mass of 200 kDa and its active state is a homodimer.[21] PDE5 consists of monomers and each contains two major functional domains: the regulatory domain (R domain) which is located in the N-terminal portion of the protein and the catalytic domain (C domain) located in the more C-terminal portion of the protein.[33][21]
The R domain contains specific allosteric cGMP binding site that controls the enzymes function. This specific binding site consists of subdomain GAF (cGMP-specific cGMP-stimulated PDE, adenylate cyclase, and FhlA) which is located in the N-terminal section of the specific proteins. The allosteric binding site GAF consists of GAFa and GAFb where GAFa has a higher binding affinity. The importance and functional role of the two homologous binding sites are unknown.[34]
Conformational change occurs when cGMP binds to the allosteric site that exposes serine and permits phosphorylation. The results for the phosphorylation of serine leads to increased cGMP hydrolysis at the catalytic domain. The affinity of the catalytic domain for cGMP increases and further increases the PDE5 catalytic domain activity.[33] Through the C domain, intracellular cGMP is degraded rapidly by PDE5 which minimizes the activity of cGMP on its PKG1 substrate by cleaving the cyclic phosphate part of cGMP to GMP. GMP is an inactive molecule with no second messenger activity.[33][35]
Phosphorylation of a single serine by PKG1 and the allosteric cGMP binding site activates the PDE5 catalytic activity and the result is a negative feedback regulation of cGMP/NO/PKG1 signalling. cGMP therefore interacts with both allosteric and catalytic domain of the PDE5 enzyme and PDE5 inhibitors compete with cGMP for binding at the catalytic domain resulting in higher cGMP levels.[33] PDE5 domains are demonstrated in figure 2.
PDE5 Inhibitors
Figure 3. A flow chart showing the relationship between different parts (domains) of PDE5-inhibitors. Figure 4. A representation of the 3D structure of sildenafil, a common PDE5-inhibitor
The PDE5 inhibitors sildenafil, vardenafil and tadalafil are competitive and reversible inhibitors of cGMP hydrolysis by the catalytic side of PDE5. The structures of vardenafil and sildenafil are similar, they both contain similar structured purine ring of cGMP that contributes their features to act as a competitive inhibitor of PDE5. The difference of the molecular structures is the reason for interaction with the catalytic site of PDE5 and improves the affinity of these compounds compared with cGMP selectivity.[33]
Pharmacophore
The pharmacophore model of PDE5 usually consists of one hydrogen bond acceptor, one hydrophobic aliphatic carbon chain and two aromatic rings. Small hydrophobic pocket and H-loop of PDE5 enzyme are important for binding affinity of PDE5 inhibitors. As well as positional and conformational changes are observed upon inhibitor binding in many cases.[36]
The active site of PDE5 is located at a helical bundle domain at the center of C domain (catalytic domain). The substrate pocket is composed of four subsites: M site (metal-binding site), Q pocket (core pocket), H pocket (hydrophobic pocket) and L region (lid region) as demonstrated in figure 3.[37] The Q pocket accommodates the pyrazolopyrimidinone group of sildenafil. That suggest that other chemicals similar to guanidine groups of cGMP can also bind at this region. The amino acids residues, Gln817, Phe820, Val782 and Tyr612, are lined in the Q pocket, they are highly conserved in all PDEs. The amide moiety of the pyrazolopyrimidinone group forms a bidentate hydrogen bond with the ɣ-amide group of Gln817.[37] 3D structure of sildenafil is demonstrated in figure 4.
Side effects
PDE5 inhibitors are generally well tolerated, with side effects including transient headaches, flushing, dyspepsia, congestion and dizziness.[3] There have also been reports of temporary vision disturbances with sildenafil and, to a lesser extent, vardenafil, and back and muscle pain with tadalafil.[3] These side effects may be attributed to the unintended effects of PDE5 inhibitors against other PDE isozymes, such as PDE1, PDE6 and PDE11. It is theorised that improved selectivity of PDE5 inhibitors may lead to fewer side effects.[3] For example, vardenafil and tadalafil have demonstrated reduced adverse effects probably due to improved selectivity for PDE5.[38] However, no highly selective PDE5 inhibitors are currently in development.[3]
Patients who take nitrates, alpha blockers or sGC stimulators within 24 hours of PDE5 inhibitor administration (or 48 hours for tadalafil) may experience symptomatic hypotension, so concurrent use is contraindicated.[3] PDE5 inhibitors are also contraindicated in patients with hereditary eye conditions such as retinitis pigmentosa due to the small increased risk of nonarteritic ischaemic optic neuropathy in patients taking the medication.[3]
Hearing impairment is one risk factor for those who are using PDE5 inhibitors and it has been reported for all available drugs on the market. This problem may be due to high level effect cGMP on cochlear hair cells.[33] It has been reported that PDE5 inhibitors (sildenafil & vardenafil) cause transient visual disturbances likely due to PDE6 inhibition.[3]
Several reports are about approaches to improve PDE5 inhibitors, where as chemical groups have been switched out to increase potency and selectivity, which should potentially lead to drugs with fewer side effects.[38][39]
Structure–activity relationship (SAR)
Sildenafil, the first PDE5 inhibitor, was discovered through rational drug design programme. The compound was potent and selective over PDE5 but was lacking preferable pharmacological properties.[40]
Structure-activity relationship (SAR) is demonstrated in figure 5, figure 6 and figure 7. Figure 5 demonstrates the three main groups of sildenafil, R1, R2 and R3. R1 is the pyrazolopyrimidinone ring, R2 the ethoxyphenyl ring and R3 is the methylpiperazine ring. R1 group is responsible for the binding of the drug to its active binding site of PDE5.[27]
Figure 5. PDE5 SAR1
Solubility is one of the pharmacological properties that was increased. A group was substituted for the hydrogen atom as demonstrated in figure 6. The sulfonamide group was chosen to lower lipophilicity and increase solubility as seen in figure 7.[1][39]
Figure 6. PDE5 SAR2
Solubility was further increased by placing a methyl group at R positions as demonstrated in figure 7. Other phosphodiesterase-5 inhibitors were developed from the structure in figure 7.[1][39]
Figure 7. PDE5 SAR3
Other research
Although PDE5 inhibitors main use has been for erectile dysfunction there has been a great interest in PDE5 inhibitors as a promising new therapeutic agents for treatment of other diseases, such as Alzheimer's disease. Elevation of cGMP levels through inhibition of PDE5 provides a way of improving memory and learning.[1] PDE5 has also been considered as a potential therapeutic agent for parasitic disease such as African sleeping sickness. Strategic changes were made to the structure of sildenafil so the molecule could project into a parasite-specific pocket (the p-pocket). Similar approach has been used to design therapeutic agents Plasmodium falciparum.[2]
12Bingham, J.; Sudarsanam, S. & Srinivasan, S. (2006). "Profiling human phosphodiesterase genes and splice isoforms". Biochemical and Biophysical Research Communications 350, 25-32.
12Jiang, W. Q.; etal. (2004). "Profiling Synthesis and SAR of tetracyclic pyrroloquinolones as phosphodiesterase 5 inhibitors". Bioorganic & Medicinal Chemistry 12, 1505-1515.
↑Garrett (2002). Principles of biochemistry: with a human focus. Fort Worth: Harcourt College Publishers. ISBN978-0-03-097369-7.
1234567Ghofrani, H. A.; Osterloh, I. H. & Grimminger, F. (2006). "Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond". Nature Reviews Drug Discovery 5, 689-702.
12Sung, B. J.; etal. (2003). "Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules". Nature 425, 98-102.
123456789101112Jeon, Y. H.; etal. (2005). "Phosphodiesterase: overview of protein structures, potential therapeutic applications and recent progress in drug development". Cmls-Cellular and Molecular Life Sciences 62, 1198-1220.
12Lin, C. S. (2004). "Tissue expression, distribution, and regulation of PDE5". International Journal of Impotence Research 16, S8-S10.
12345Jackson, G.; Gillies, H. & Osterloh, I. (2005). "Past, present, and future: a 7-year update of Viagra((R)) (sildenafil citrate)". International Journal of Clinical Practice 59, 680-691.
↑Blount, M. A.; etal. (2004). "Binding of tritiated sildenafil, tadalafil, or vardenafil to the phosphodiesterase-5 catalytic site displays potency, specificity, heterogeneity, and cGMP stimulation". Molecular Pharmacology 66, 144-152.
↑Lugnier, C. (2006). "Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents". Pharmacology & Therapeutics 109, 366-398.
1234567Card, G. L.; etal. (2004). "Structural basis for the activity of drugs that inhibit phosphodiesterases". Structure 12, 2233-2247.
12McMahon, C. G.; McMahon, C. N.; Leow, L. J. & Winestock, C. G. (2006). "Efficacy of type-5 phosphodiesterase inhibitors in the drug treatment of premature ejaculation: a systematic review". Bju International 98, 259-272.
↑Shinlapawittayatorn, K.; Chattipakorn, S. & Chattipakorn, N. (2005). "Effect of sildenafil citrate on the cardiovascular system". Brazilian Journal of Medical and Biological Research 38, 1303-1311.
↑Chung, K. F. (2006). "Phosphodiesterase inhibitors in airways disease". European Journal of Pharmacology 533, 110-117.
1234Rotella, D. P. (2002). "Phosphodiesterase 5 inhibitors: current status and potential applications". Nature Reviews. Drug Discovery. 1 (9): 674–682. doi:10.1038/nrd893. PMID12209148. S2CID11807377.
123Ravipati, G.; McClung, J. A.; Aronow, W. S.; Peterson, S. J.; Frishman, W. H. (2007). "Type 5 phosphodiesterase inhibitors in the treatment of erectile dysfunction and cardiovascular disease". Cardiol Rev. 15 (2): 76–86. doi:10.1097/01.crd.0000233904.77128.49. PMID17303994. S2CID23461685.
12Beer, D.; Bhalay, G.; Dunstan, A.; Glen, A.; Haberthuer, S.; Moser, H.; Jiang, H. (2002). "A solid-phase approach towards the synthesis of PDE5 inhibitors". Bioorg Med Chem Lett. 12 (15): 1973–1976. doi:10.1016/S0960-894X(02)00296-2. PMID12113821.
12Yu, G. X.; Mason, H.; Wu, X. M.; Wang, J.; Chong, S. H.; Beyer, B. (2003). "Substituted pyrazolopyridopyridazines as orally bioavailable potent and selective PDE5 inhibitors: Potential agents for treatment of erectile dysfunction". J Med Chem. 46 (4): 457–460. doi:10.1021/Jm0256068. PMID12570368.
123Pissarnitski, D. A.; Asberom, T.; Boyle, C. D.; Chackalamannil, S.; Chintala, M.; Clader, J. W.; Xu, R. (2004). "SAR development of polycyclic guanine derivatives targeted to the discovery of a selective PDE5 inhibitor for treatment of erectile dysfunction". Bioorg Med Chem Lett. 14 (5): 1291–1294. doi:10.1016/j.bmcl.2003.12.027. PMID14980684.
↑Campbell, S.F. (2000). "Science, art and drug discovery: a personal perspective". Clinical Science. 99 (4): 255–260. doi:10.1042/cs20000140. PMID10995589.
↑Yu, J. Y.; Kang, K. K. & Yoo, M. (2006). "Erectile potentials of a new phosphodiesterase type 5 inhibitor, DA-8159, in diet-induced obese rats". Asian Journal of Andrology 8, 325-329.
↑Ahn, G. J.; etal. (2005). "Chronic administration of phosphodiesterase 5 inhibitor improves erectile and endothelial function in a rat model of diabetes". International Journal of Andrology 28, 260-266.
↑Ahn, G. J.; etal. (2005). "DA-8159 reverses selective serotonin reuptake inhibitor-induced erectile dysfunction in rats". Urology 65, 202-207.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.










{kind=link}
{kind=link}