Calcium channel blockers (CCB), calcium channel antagonists or calcium antagonists[2] are a group of medications that disrupt the movement of calcium (Ca2+) through calcium channels.[3] Calcium channel blockers are used as antihypertensive drugs, i.e., as medications to decrease blood pressure in patients with hypertension. CCBs are particularly effective against large vessel stiffness, one of the common causes of elevated systolic blood pressure in elderly patients.[4] Calcium channel blockers are also frequently used to alter heart rate (especially from atrial fibrillation), to prevent peripheral and cerebral vasospasm, and to reduce chest pain caused by angina pectoris.
CCBs have been shown to be slightly more effective than beta blockers at lowering cardiovascularmortality associated with stroke, but they are associated with more side effects.[6][7] Potential major risks however were mainly found to be associated with short-acting CCBs.[8]
Classes
Dihydropyridine
General chemical structure of dihydropyridine calcium channel blockers (dipines)
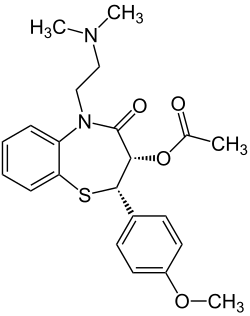
Dihydropyridine (DHP) calcium channel blockers are derived from the molecule dihydropyridine and often used to reduce systemic vascular resistance and arterial pressure. Sometimes when they are used to treat angina, the vasodilation and hypotension can lead to reflex tachycardia, which can be detrimental for patients with ischemic symptoms because of the resulting increase in myocardial oxygen demand. Dihydropyridine calcium channel blockers can worsen proteinuria in patients with nephropathy.[9]
This CCB class is easily identified by the suffix "-dipine".
Phenylalkylamine calcium channel blockers are relatively selective for myocardium, reduce myocardial oxygen demand and reverse coronary vasospasm, and are often used to treat angina. They have minimal vasodilatory effects compared with dihydropyridines and therefore cause less reflex tachycardia, making it appealing for treatment of angina, where tachycardia can be the most significant contributor to the heart's need for oxygen. Therefore, as vasodilation is minimal with the phenylalkylamines, the major mechanism of action is causing negative inotropy. Phenylalkylamines are thought to access calcium channels from the intracellular side, although the evidence is somewhat mixed.[10]
Benzothiazepine calcium channel blockers belong to the benzothiazepine class of compounds and are an intermediate class between phenylalkylamine and dihydropyridines in their selectivity for vascular calcium channels. By having both cardiac depressant and vasodilator actions, benzothiazepines are able to reduce arterial pressure without producing the same degree of reflex cardiac stimulation caused by dihydropyridines.[citation needed]
Gabapentinoids, such as gabapentin and pregabalin, bind selectively to the α2δ protein that was first described as an integral part of voltage-gated calcium channels. These drugs do not directly block calcium channels[13][14] but can alter the transport of functional calcium channels to the cell membrane and they also reduce the release of certain excitatory neurotransmitters. They are used primarily to treat epilepsy and neuropathic pain.[15] More recently, the α2δ-1 protein has been found to bind directly and to interact with certain glutamate receptors and to the interstitial protein thrombospondin, independently from their action at calcium channels.[citation needed]
Ziconotide, a peptide compound derived from the omega-conotoxin, is a selective N-type calcium channel blocker that has potent analgesic properties that are equivalent to approximate 1,000 times that of morphine. It must be delivered via the intrathecal (directly into the cerebrospinal fluid) route via an intrathecal infusion pump.[16]
Naturally occurring compounds and elements such as magnesium have also been shown to act as calcium channel blockers when administered orally.[17]
Side effects
Side effects of these drugs may include but are not limited to:
Mild CCB toxicity is treated with supportive care. Nondihydropyridine CCBs may produce profound toxicity, and early decontamination, especially for slow-release agents, is essential. For severe overdoses, treatment usually includes close monitoring of vital signs and the addition of vasopressive agents and intravenous fluids for blood pressure support. Intravenous calcium gluconate (or calcium chloride if a central line is available) and atropine are first-line therapies. If the time of the overdose is known and presentation is within two hours of ingestion, activated charcoal, gastric lavage, and polyethylene glycol may be used to decontaminate the gut. Efforts for gut decontamination may be extended to within 8 hours of ingestion with extended-release preparations.[citation needed]
Hyperinsulinemia-euglycemia therapy has emerged as a viable form of treatment.[24] Although the mechanism is unclear, increased insulin may mobilize glucose from peripheral tissues to serve as an alternative fuel source for the heart (the heart mainly relies on oxidation of fatty acids). Theoretical treatment with lipid emulsion therapy has been considered in severe cases, but is not yet standard of care.
Caution should be taken when using verapamil with a beta blocker due to the risk of severe bradycardia. If unsuccessful, ventricular pacing should be used.[25]
Non-medical calcium channel inhibitors
Ethanol
Ethanol blocks voltage-gated calcium channel
Research indicates ethanol is involved in the inhibition of L-type calcium channels. One study showed the nature of ethanol binding to L-type calcium channels is according to first-order kinetics with a Hill coefficient around 1. This indicates ethanol binds independently to the channel, expressing noncooperative binding.[26] Early studies showed a link between calcium and the release of vasopressin by the secondary messenger system.[27] Vasopressin levels are reduced after the ingestion of alcohol.[28] The lower levels of vasopressin from the consumption of alcohol have been linked to ethanol acting as an antagonist to voltage-gated calcium channels (VGCCs). Studies conducted by Treistman et al. in the aplysia confirm inhibition of VGCC by ethanol. Voltage clamp recordings have been done on the aplysia neuron. VGCCs were isolated and calcium current was recorded using patch clamp technique having ethanol as a treatment. Recordings were replicated at varying concentrations (0, 10, 25, 50, and 100 mM) at a voltage clamp of +30 mV. Results showed calcium current decreased as concentration of ethanol increased.[29] Similar results have shown to be true in single-channel recordings from isolated nerve terminal of rats that ethanol does in fact block VGCCs.[30]
Studies done by Katsura et al. in 2006 on mouse cerebral cortical neurons, show the effects of prolonged ethanol exposure. Neurons were exposed to sustained ethanol concentrations of 50 mM for 3 days in vitro. Western blot and protein analysis were conducted to determine the relative amounts of VGCC subunit expression. α1C, α1D, and α2/δ1 subunits showed an increase of expression after sustained ethanol exposure. However, the β4 subunit showed a decrease. Furthermore, α1A, α1B, and α1F subunits did not alter in their relative expression. Thus, sustained ethanol exposure may participate in the development of ethanol dependence in neurons.[31]
Other experiments done by Malysz et al. have looked into ethanol effects on voltage-gated calcium channels on detrusor smooth muscle cells in guinea pigs. Perforated patch clamp technique was used having intracellular fluid inside the pipette and extracellular fluid in the bath with added 0.3% vol/vol (about 50-mM) ethanol. Ethanol decreased the Ca2+ current in DSM cells and induced muscle relaxation. Ethanol inhibits VGCCs and is involved in alcohol-induced relaxation of the urinary bladder.[32]
Agatoxin in spider venom
Research on the desert grass spider, Agelenopsis aperta, has shown that agatoxins IVA and IVB found in their venom selectively block calcium channels. These agatoxins are found in other spider species as well. Desert grass spider bites to insects result in rapid paralysis, but bites to humans are not considered medically significant.[33]
Mechanism of action
A calcium channel embedded in a cell membrane.
In the body's tissues, the concentration of calcium ions (Ca2+) outside cells is normally about 10,000-fold higher than the concentration inside cells. Embedded in the membrane of some cells are calcium channels. When these cells receive a certain signal, the channels open, letting calcium rush into the cell. The resulting increase in intracellular calcium has different effects in different types of cells. Calcium channel blockers prevent or reduce the opening of these channels and thereby reduce these effects.[citation needed]
Several types of calcium channels occur, with a number of classes of blockers, but almost all of them preferentially or exclusively block the L-type voltage-gated calcium channel.[34]
By acting on vascular smooth muscle, they reduce contraction of the arteries and cause an increase in arterial diameter, a phenomenon called vasodilation (CCBs do not work on venous smooth muscle).
By acting on cardiac muscles (myocardium), they reduce the force of contraction of the heart.
By slowing down the conduction of electrical activity within the heart, they slow down the heart beat.
By blocking the calcium signal on adrenal cortex cells, they directly reduce aldosterone production, which correlates to lower blood pressure.
Since blood pressure is in intimate feedback with cardiac output and peripheral resistance, with relatively low blood pressure, the afterload on the heart decreases; this decreases how hard the heart must work to eject blood into the aorta, so the amount of oxygen required by the heart decreases accordingly. This can help ameliorate symptoms of ischaemic heart disease such as angina pectoris.
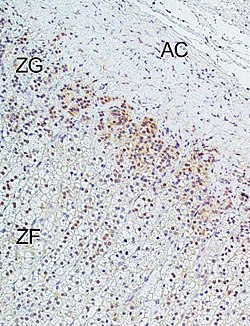
Immunohistochemical analysis of L-type calcium channel Cav1.3 (CACNA1D) in human adrenal cortex: Marked immunoreactivity was detected in the zona glomerulosa. In the figure: ZG = zona glomerulosa, ZF = zona fasciculata, AC = adrenal capsule. Immunohistochemistry was performed according to published methods.
Reducing the force of contraction of the myocardium is known as the negative inotropic effect of calcium channel blockers.
Slowing down the conduction of electrical activity within the heart, by blocking the calcium channel during the plateau phase of the action potential of the heart (see: cardiac action potential), results in a negative chronotropic effect, or a lowering of heart rate. This can increase the potential for heart block. The negative chronotropic effects of CCBs make them a commonly used class of agents in individuals with atrial fibrillation or flutter in whom control of the heart rate is generally a goal. Negative chronotropy can be beneficial when treating a variety of disease processes because lower heart rates represent lower cardiac oxygen requirements. Elevated heart rate can result in significantly higher "cardiac work", which can result in symptoms of angina.
The class of CCBs known as dihydropyridines mainly affect arterial vascular smooth muscle and lower blood pressure by causing vasodilation. The phenylalkylamine class of CCBs mainly affect the cells of the heart and have negative inotropic and negative chronotropic effects. The benzothiazepine class of CCBs combine effects of the other two classes.
Because of the negative inotropic effects, the nondihydropyridine calcium channel blockers should be avoided (or used with caution) in individuals with cardiomyopathy.[35]
Unlike beta blockers, calcium channel blockers do not decrease the responsiveness of the heart to input from the sympathetic nervous system. Since moment-to-moment blood pressure regulation is carried out by the sympathetic nervous system (via the baroreceptor reflex), calcium channel blockers allow blood pressure to be maintained more effectively than do beta blockers. However, because dihydropyridine CCBs result in a decrease in blood pressure, the baroreceptor reflex often initiates a reflexive increase in sympathetic activity leading to increased heart rate and contractility.
Ionic calcium is antagonized by magnesium ions in the nervous system. Because of this, bioavailable supplements of magnesium, possibly including magnesium chloride, magnesium lactate, and magnesium aspartate, may increase or enhance the effects of calcium channel blockade.[36]
Calcium channel blockers came into wide use in the 1960s,[37] having been first identified in the lab of German pharmacologist Albrecht Fleckenstein in 1964.[38]
123Felizola SJ, Maekawa T, Nakamura Y, Satoh F, Ono Y, Kikuchi K, Aritomi S, Ikeda K, Yoshimura M, Tojo K, Sasano H (2014). "Voltage-gated calcium channels in the human adrenal and primary aldosteronism". J Steroid Biochem Mol Biol. 144 (part B): 410–16. doi:10.1016/j.jsbmb.2014.08.012. PMID25151951. S2CID23622821.
↑Remuzzi G, Scheppati A, Ruggenenti P (2002). "Clinical Practice. Nephropathy in Patients with Type 2 Diabetes". New England Journal of Medicine. 346 (15): 1145–51. doi:10.1056/NEJMcp011773. PMID11948275.
↑Hockerman, G.H., Peterson, B.Z., Johnson, B.D., Catterall, W.A. (1997). "Molecular Determinants of Drug Binding and Action on L-Type Calcium Channels". Annual Review of Pharmacology and Toxicology. 37: 361–96. doi:10.1146/annurev.pharmtox.37.1.361. PMID9131258. S2CID16275155.
12Mohanakumar S, Telinius N, Kelly B, Hjortdal V (2019-08-20). "Reduced Lymphatic Function Predisposes to Calcium Channel Blocker Edema: A Randomized Placebo-Controlled Clinical Trial". Lymphatic Research and Biology. 18 (2). Mary Ann Liebert Inc: 156–165. doi:10.1089/lrb.2019.0028. ISSN1539-6851. PMID31429625. S2CID201094829.
↑Chiodera P, Coiro V (May 1990). "Inhibitory effect of ethanol on the arginine vasopressin response to insulin-induced hypoglycemia and the role of endogenous opioids". Neuroendocrinology. 51 (5): 501–04. doi:10.1159/000125383. PMID2112727.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.