
A primer is a short single-stranded nucleic acid used by all living organisms in the initiation of DNA synthesis. DNA polymerase enzymes are only capable of adding nucleotides to the 3’-end of an existing nucleic acid, requiring a primer be bound to the template before DNA polymerase can begin a complementary strand. DNA polymerase adds nucleotides after binding to the RNA primer and synthesizes the whole strand. Later, the RNA strands must be removed accurately and replace them with DNA nucleotides forming a gap region known as a nick that is filled in using an enzyme called ligase. The removal process of the RNA primer requires several enzymes, such as Fen1, Lig1, and others that work in coordination with DNA polymerase, to ensure the removal of the RNA nucleotides and the addition of DNA nucleotides. Living organisms use solely RNA primers, while laboratory techniques in biochemistry and molecular biology that require in vitro DNA synthesis usually use DNA primers, since they are more temperature stable. Primers can be designed in laboratory for specific reactions such as polymerase chain reaction (PCR). When designing PCR primers, there are specific measures that must be taken into consideration, like the melting temperature of the primers and the annealing temperature of the reaction itself. Moreover, the DNA binding sequence of the primer in vitro has to be specifically chosen, which is done using a method called basic local alignment search tool (BLAST) that scans the DNA and finds specific and unique regions for the primer to bind.

A Southern blot is a method used in molecular biology for detection of a specific DNA sequence in DNA samples. Southern blotting combines transfer of electrophoresis-separated DNA fragments to a filter membrane and subsequent fragment detection by probe hybridization.
Oligonucleotides are short DNA or RNA molecules, oligomers, that have a wide range of applications in genetic testing, research, and forensics. Commonly made in the laboratory by solid-phase chemical synthesis, these small bits of nucleic acids can be manufactured as single-stranded molecules with any user-specified sequence, and so are vital for artificial gene synthesis, polymerase chain reaction (PCR), DNA sequencing, molecular cloning and as molecular probes. In nature, oligonucleotides are usually found as small RNA molecules that function in the regulation of gene expression, or are degradation intermediates derived from the breakdown of larger nucleic acid molecules.

Ribonuclease is a type of nuclease that catalyzes the degradation of RNA into smaller components. Ribonucleases can be divided into endoribonucleases and exoribonucleases, and comprise several sub-classes within the EC 2.7 and 3.1 classes of enzymes.

A nuclease is an enzyme capable of cleaving the phosphodiester bonds between nucleotides of nucleic acids. Nucleases variously effect single and double stranded breaks in their target molecules. In living organisms, they are essential machinery for many aspects of DNA repair. Defects in certain nucleases can cause genetic instability or immunodeficiency. Nucleases are also extensively used in molecular cloning.

Ribonuclease H is a family of non-sequence-specific endonuclease enzymes that catalyze the cleavage of RNA in an RNA/DNA substrate via a hydrolytic mechanism. Members of the RNase H family can be found in nearly all organisms, from bacteria to archaea to eukaryotes.

Okazaki fragments are short sequences of DNA nucleotides which are synthesized discontinuously and later linked together by the enzyme DNA ligase to create the lagging strand during DNA replication. They were discovered in the 1960s by the Japanese molecular biologists Reiji and Tsuneko Okazaki, along with the help of some of their colleagues.
A cDNA library is a combination of cloned cDNA fragments inserted into a collection of host cells, which constitute some portion of the transcriptome of the organism and are stored as a "library". cDNA is produced from fully transcribed mRNA found in the nucleus and therefore contains only the expressed genes of an organism. Similarly, tissue-specific cDNA libraries can be produced. In eukaryotic cells the mature mRNA is already spliced, hence the cDNA produced lacks introns and can be readily expressed in a bacterial cell. While information in cDNA libraries is a powerful and useful tool since gene products are easily identified, the libraries lack information about enhancers, introns, and other regulatory elements found in a genomic DNA library.

Exodeoxyribonuclease V is an enzyme of E. coli that initiates recombinational repair from potentially lethal double strand breaks in DNA which may result from ionizing radiation, replication errors, endonucleases, oxidative damage, and a host of other factors. The RecBCD enzyme is both a helicase that unwinds, or separates the strands of DNA, and a nuclease that makes single-stranded nicks in DNA. It catalyses exonucleolytic cleavage in either 5′- to 3′- or 3′- to 5′-direction to yield 5′-phosphooligonucleotides.
Endonucleases are enzymes that cleave the phosphodiester bond within a polynucleotide chain. Some, such as deoxyribonuclease I, cut DNA relatively nonspecifically, while many, typically called restriction endonucleases or restriction enzymes, cleave only at very specific nucleotide sequences. Endonucleases differ from exonucleases, which cleave the ends of recognition sequences instead of the middle (endo) portion. Some enzymes known as "exo-endonucleases", however, are not limited to either nuclease function, displaying qualities that are both endo- and exo-like. Evidence suggests that endonuclease activity experiences a lag compared to exonuclease activity.

Exonucleases are enzymes that work by cleaving nucleotides one at a time from the end (exo) of a polynucleotide chain. A hydrolyzing reaction that breaks phosphodiester bonds at either the 3′ or the 5′ end occurs. Its close relative is the endonuclease, which cleaves phosphodiester bonds in the middle (endo) of a polynucleotide chain. Eukaryotes and prokaryotes have three types of exonucleases involved in the normal turnover of mRNA: 5′ to 3′ exonuclease (Xrn1), which is a dependent decapping protein; 3′ to 5′ exonuclease, an independent protein; and poly(A)-specific 3′ to 5′ exonuclease.
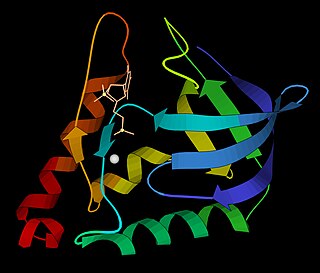
Micrococcal nuclease is an endo-exonuclease that preferentially digests single-stranded nucleic acids. The rate of cleavage is 30 times greater at the 5' side of A or T than at G or C and results in the production of mononucleotides and oligonucleotides with terminal 3'-phosphates. The enzyme is also active against double-stranded DNA and RNA and all sequences will be ultimately cleaved.
Nuclease protection assay is a laboratory technique used in biochemistry and genetics to identify individual RNA molecules in a heterogeneous RNA sample extracted from cells. The technique can identify one or more RNA molecules of known sequence even at low total concentration. The extracted RNA is first mixed with antisense RNA or DNA probes that are complementary to the sequence or sequences of interest and the complementary strands are hybridized to form double-stranded RNA. The mixture is then exposed to ribonucleases that specifically cleave only single-stranded RNA but have no activity against double-stranded RNA. When the reaction runs to completion, susceptible RNA regions are degraded to very short oligomers or to individual nucleotides; the surviving RNA fragments are those that were complementary to the added antisense strand and thus contained the sequence of interest.

Systematic evolution of ligands by exponential enrichment (SELEX), also referred to as in vitro selection or in vitro evolution, is a combinatorial chemistry technique in molecular biology for producing oligonucleotides of either single-stranded DNA or RNA that specifically bind to a target ligand or ligands. These single-stranded DNA or RNA are commonly referred to as aptamers. Although SELEX has emerged as the most commonly used name for the procedure, some researchers have referred to it as SAAB and CASTing SELEX was first introduced in 1990. In 2015, a special issue was published in the Journal of Molecular Evolution in the honor of quarter century of the discovery of SELEX.
This glossary of genetics is a list of definitions of terms and concepts commonly used in the study of genetics and related disciplines in biology, including molecular biology, cell biology, and evolutionary biology. It is intended as introductory material for novices; for more specific and technical detail, see the article corresponding to each term. For related terms, see Glossary of evolutionary biology.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.

Nuclease S1 is an endonuclease enzyme that splits single-stranded DNA (ssDNA) and RNA into oligo- or mononucleotides. This enzyme catalyses the following chemical reaction
Deoxyribonuclease IV (phage-T4-induced) is catalyzes the degradation nucleotides in DsDNA by attacking the 5'-terminal end.
A hybridization assay comprises any form of quantifiable hybridization i.e. the quantitative annealing of two complementary strands of nucleic acids, known as nucleic acid hybridization.
SCAR-less genome editing Scarless Cas9 Assisted Recombineering (no-SCAR) is an editing method that is able to manipulate the Escherichia coli genome. The system relies on recombineering whereby DNA sequences are combined and manipulated through homologous recombination. No-SCAR is able to manipulate the E. coli genome without the use of the chromosomal markers detailed in previous recombineering methods. Instead, in this method, the λ-Red recombination system facilitates donor DNA integration while Cas9 cleaves double-stranded DNA to counter-select against wild-type cells. Although λ-Red and Cas9 genome editing are widely used technologies, the no-SCAR method is novel in combining the two functions; this technique is able to establish point mutations, gene deletions, and short sequence insertions in several genomic loci with increased efficiency and time sensitivity.