Antiandrogens, also known as androgen antagonists or testosterone blockers, are a class of drugs that prevent androgens like testosterone and dihydrotestosterone (DHT) from mediating their biological effects in the body. They act by blocking the androgen receptor (AR) and/or inhibiting or suppressing androgen production. They can be thought of as the functional opposites of AR agonists, for instance androgens and anabolic steroids (AAS) like testosterone, DHT, and nandrolone and selective androgen receptor modulators (SARMs) like enobosarm. Antiandrogens are one of three types of sex hormone antagonists, the others being antiestrogens and antiprogestogens.
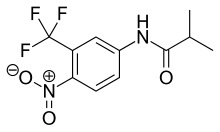
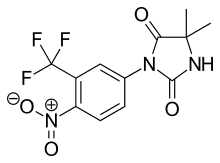
Bicalutamide, sold under the brand name Casodex among others, is an antiandrogen medication that is primarily used to treat prostate cancer. It is typically used together with a gonadotropin-releasing hormone (GnRH) analogue or surgical removal of the testicles to treat metastatic prostate cancer (mPC). To a lesser extent, it is used at high doses for locally advanced prostate cancer (LAPC) as a monotherapy without castration. Bicalutamide was also previously used as monotherapy to treat localized prostate cancer (LPC), but authorization for this use was withdrawn following unfavorable trial findings. Besides prostate cancer, bicalutamide is limitedly used in the treatment of excessive hair growth and scalp hair loss in women, as a puberty blocker and component of feminizing hormone therapy for transgender girls and women, to treat gonadotropin-independent early puberty in boys, and to prevent overly long-lasting erections in men. It is taken by mouth.

Flutamide, sold under the brand name Eulexin among others, is a nonsteroidal antiandrogen (NSAA) which is used primarily to treat prostate cancer. It is also used in the treatment of androgen-dependent conditions like acne, excessive hair growth, and high androgen levels in women. It is taken by mouth, usually three times per day.

Nilutamide, sold under the brand names Nilandron and Anandron, is a nonsteroidal antiandrogen (NSAA) which is used in the treatment of prostate cancer. It has also been studied as a component of feminizing hormone therapy for transgender women and to treat acne and seborrhea in women. It is taken by mouth.

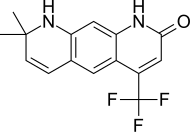
Enzalutamide, sold under the brand name Xtandi, is a nonsteroidal antiandrogen (NSAA) medication which is used in the treatment of prostate cancer. It is indicated for use in conjunction with castration in the treatment of metastatic castration-resistant prostate cancer (mCRPC), nonmetastatic castration-resistant prostate cancer, and metastatic castration-sensitive prostate cancer (mCSPC). It is taken by mouth.

Cyproterone, also known by its developmental code name SH-80881, is a steroidal antiandrogen which was studied in the 1960s and 1970s but was never introduced for medical use. It is a precursor of cyproterone acetate (CPA), an antiandrogen, progestin, and antigonadotropin which was introduced instead of cyproterone and is widely used as a medication. Cyproterone and CPA were among the first antiandrogens to be developed.
EPI-001 is the first inhibitor of the androgen receptor amino-terminal domain. The single stereoisomer of EPI-001, EPI-002, is a first-in-class drug that the USAN council assigned a new stem class "-aniten" and the generic name "ralaniten". This distinguishes the anitens novel molecular mechanism from anti androgens that bind the C-terminus ligand-binding domain and have the stem class "lutamide". EPI-001 and its stereoisomers and analogues were discovered by Marianne Sadar and Raymond Andersen, who co-founded the pharmaceutical company ESSA Pharma Inc for the clinical development of anitens for the treatment of castration-resistant prostate cancer (CRPC).

Benorterone, also known by its developmental code name SKF-7690 and as 17α-methyl-B-nortestosterone, is a steroidal antiandrogen which was studied for potential medical use but was never marketed. It was the first known antiandrogen to be studied in humans. It is taken by mouth or by application to skin.

Hydroxyflutamide (HF, OHF) (developmental code name SCH-16423), or 2-hydroxyflutamide, is a nonsteroidal antiandrogen (NSAA) and the major active metabolite of flutamide, which is considered to be a prodrug of hydroxyflutamide as the active form. It has been reported to possess an IC50 of 700 nM for the androgen receptor (AR), which is about 4-fold less than that of bicalutamide.
A nonsteroidal antiandrogen (NSAA) is an antiandrogen with a nonsteroidal chemical structure. They are typically selective and full or silent antagonists of the androgen receptor (AR) and act by directly blocking the effects of androgens like testosterone and dihydrotestosterone (DHT). NSAAs are used in the treatment of androgen-dependent conditions in men and women. They are the converse of steroidal antiandrogens (SAAs), which are antiandrogens that are steroids and are structurally related to testosterone.

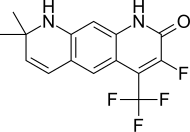
Apalutamide, sold under the brand name Erleada among others, is a nonsteroidal antiandrogen (NSAA) medication used for the treatment of prostate cancer. It is an androgen receptor inhibitor. It is taken by mouth.

BOMT, also known by its developmental code name Ro 7-2340 and as 6α-bromo-4-oxa-17α-methyl-5α-dihydrotestosterone, is a synthetic steroidal antiandrogen which was first produced in 1970 and was never marketed for medical use. It is the 6α-brominated, 4-oxygenated, and 17α-methylated derivative of the androgen dihydrotestosterone (DHT). Along with benorterone, cyproterone, and flutamide, BOMT was among the earliest antiandrogens to be developed and extensively studied, although it is less well-documented in comparison to the others. BOMT has been investigated clinically in the treatment of benign prostatic hyperplasia, though development for this use did not continue. There was also interest in BOMT for the potential applications of acne, pattern hair loss, and possibly prostate cancer, but it was not developed for these indications either.

Trimethyltrienolone (TMT), also known by its developmental code name R-2956 or RU-2956, is an antiandrogen medication which was never introduced for medical use but has been used in scientific research.
The medical uses of bicalutamide, a nonsteroidal antiandrogen (NSAA), include the treatment of androgen-dependent conditions and hormone therapy to block the effects of androgens. Indications for bicalutamide include the treatment of prostate cancer in men, skin and hair conditions such as acne, seborrhea, hirsutism, and pattern hair loss in women, high testosterone levels in women, hormone therapy in transgender women, as a puberty blocker to prevent puberty in transgender girls and to treat early puberty in boys, and the treatment of long-lasting erections in men. It may also have some value in the treatment of paraphilias and hypersexuality in men.
Comparison of the nonsteroidal antiandrogen (NSAA) bicalutamide with other antiandrogens reveals differences between the medications in terms of efficacy, tolerability, safety, and other parameters. Relative to the other first-generation NSAAs, flutamide and nilutamide, bicalutamide shows improved potency, efficacy, tolerability, and safety, and has largely replaced these medications in clinical practice. Compared to the second-generation NSAAs, enzalutamide and apalutamide, bicalutamide has inferior potency and efficacy but similar tolerability and safety and a lower propensity for drug interactions.
The pharmacology of bicalutamide is the study of the pharmacodynamic and pharmacokinetic properties of the nonsteroidal antiandrogen (NSAA) bicalutamide. In terms of pharmacodynamics, bicalutamide acts as a selective antagonist of the androgen receptor (AR), the biological target of androgens like testosterone and dihydrotestosterone (DHT). It has no capacity to activate the AR. It does not decrease androgen levels and has no other important hormonal activity. The medication has progonadotropic effects due to its AR antagonist activity and can increase androgen, estrogen, and neurosteroid production and levels. This results in a variety of differences of bicalutamide monotherapy compared to surgical and medical castration, such as indirect estrogenic effects and associated benefits like preservation of sexual function and drawbacks like gynecomastia. Bicalutamide can paradoxically stimulate late-stage prostate cancer due to accumulated mutations in the cancer. When used as a monotherapy, bicalutamide can induce breast development in males due to its estrogenic effects. Unlike other kinds of antiandrogens, it may have less adverse effect on the testes and fertility.

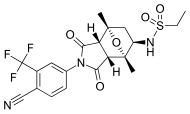
RD-162 is a second-generation nonsteroidal antiandrogen (NSAA) which was developed for the treatment of prostate cancer but was never marketed. It acts as a potent and selective silent antagonist of the androgen receptor (AR). The drug is a diarylthiohydantoin derivative. It is closely related to enzalutamide and apalutamide. Both RD-162 and enzalutamide show 5- to 8-fold higher affinity for the AR than the first-generation NSAA bicalutamide, and only 2- to 3-fold lower affinity than dihydrotestosterone (DHT), the major endogenous ligand of the receptor in the prostate gland.

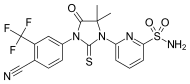
RU-59063 is a nonsteroidal androgen or selective androgen receptor modulator (SARM) which was first described in 1994 and was never marketed. It was originally thought to be a potent antiandrogen, but subsequent research found that it actually possesses dose-dependent androgenic activity, albeit with lower efficacy than dihydrotestosterone (DHT). The drug is an N-substituted arylthiohydantoin and was derived from the first-generation nonsteroidal antiandrogen (NSAA) nilutamide. The second-generation NSAAs enzalutamide, RD-162, and apalutamide were derived from RU-59063.

EM-5854 is a steroidal antiandrogen which was under development by Endoceutics, Inc. for the treatment of prostate cancer. It was first described in a patent in 2008, and was further characterized in 2012. EM-5854 reached phase I/II clinical trials for the treatment of prostate cancer but development was discontinued in March 2019.

The pharmacology of cyproterone acetate (CPA) concerns the pharmacology of the steroidal antiandrogen and progestin medication cyproterone acetate.