
Antiandrogens, also known as androgen antagonists or testosterone blockers, are a class of drugs that prevent androgens like testosterone and dihydrotestosterone (DHT) from mediating their biological effects in the body. They act by blocking the androgen receptor (AR) and/or inhibiting or suppressing androgen production. They can be thought of as the functional opposites of AR agonists, for instance androgens and anabolic steroids (AAS) like testosterone, DHT, and nandrolone and selective androgen receptor modulators (SARMs) like enobosarm. Antiandrogens are one of three types of sex hormone antagonists, the others being antiestrogens and antiprogestogens.

In the field of molecular biology, the peroxisome proliferator–activated receptors (PPARs) are a group of nuclear receptor proteins that function as transcription factors regulating the expression of genes. PPARs play essential roles in the regulation of cellular differentiation, development, and metabolism, and tumorigenesis of higher organisms.
Bicalutamide, sold under the brand name Casodex among others, is an antiandrogen medication that is primarily used to treat prostate cancer. It is typically used together with a gonadotropin-releasing hormone (GnRH) analogue or surgical removal of the testicles to treat metastatic prostate cancer (mPC). To a lesser extent, it is used at high doses for locally advanced prostate cancer (LAPC) as a monotherapy without castration. Bicalutamide was also previously used as monotherapy to treat localized prostate cancer (LPC), but authorization for this use was withdrawn following unfavorable trial findings. Besides prostate cancer, bicalutamide is limitedly used in the treatment of excessive hair growth and scalp hair loss in women, as a puberty blocker and component of feminizing hormone therapy for transgender girls and women, to treat gonadotropin-independent early puberty in boys, and to prevent overly long-lasting erections in men. It is taken by mouth.
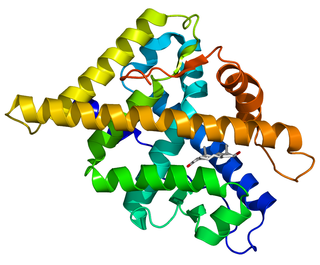
The androgen receptor (AR), also known as NR3C4, is a type of nuclear receptor that is activated by binding any of the androgenic hormones, including testosterone and dihydrotestosterone, in the cytoplasm and then translocating into the nucleus. The androgen receptor is most closely related to the progesterone receptor, and progestins in higher dosages can block the androgen receptor.

Enzalutamide, sold under the brand name Xtandi, is a nonsteroidal antiandrogen (NSAA) medication which is used in the treatment of prostate cancer. It is indicated for use in conjunction with castration in the treatment of metastatic castration-resistant prostate cancer (mCRPC), nonmetastatic castration-resistant prostate cancer, and metastatic castration-sensitive prostate cancer (mCSPC). It is taken by mouth.
The first antiandrogen was discovered in the 1960s. Antiandrogens antagonise the androgen receptor (AR) and thereby block the biological effects of testosterone and dihydrotestosterone (DHT). Antiandrogens are important for men with hormonally responsive diseases like prostate cancer, benign prostatic hyperplasia (BHP), acne, seborrhea, hirsutism and androgen alopecia. Antiandrogens are mainly used for the treatment of prostate diseases. Research from 2010 suggests that ARs could be linked to the disease progression of triple-negative breast cancer and salivary duct carcinoma and that antiandrogens can potentially be used to treat it.

Galeterone is a steroidal antiandrogen which was under development by Tokai Pharmaceuticals for the treatment of prostate cancer. It possesses a unique triple mechanism of action, acting as an androgen receptor antagonist, androgen receptor down regulator, and CYP17A1 inhibitor, the latter of which prevents the biosynthesis of androgens. As a CYP17A1 inhibitor, galeterone shows selectivity for 17,20-lyase over 17α-hydroxylase.

Apalutamide, sold under the brand name Erleada among others, is a nonsteroidal antiandrogen (NSAA) medication used for the treatment of prostate cancer. It is an androgen receptor inhibitor. It is taken by mouth.

Ralaniten acetate is a first-in-class antiandrogen that targets the N-terminal domain (NTD) of the androgen receptor (AR) developed by ESSA Pharmaceuticals and was under investigation for the treatment of prostate cancer. This mechanism of action is believed to allow the drug to block signaling from the AR and its splice variants. EPI-506 is a derivative of bisphenol A and a prodrug of ralaniten (EPI-002), one of the four stereoisomers of EPI-001, and was developed as a successor of EPI-001. The drug reached phase I/II prior to the discontinuation of its development. It showed signs of efficacy in the form of prostatic specific antigen (PSA) decreases (4–29%) predominantly at higher doses (≥1,280 mg) in some patients but also caused side effects and was discontinued by its developer in favor of next-generation AR NTD inhibitors with improved potency and tolerability.
A hormone-sensitive cancer, or hormone-dependent cancer, is a type of cancer that is dependent on a hormone for growth and/or survival. Examples include breast cancer, which is dependent on estrogens like estradiol, and prostate cancer, which is dependent on androgens like testosterone.
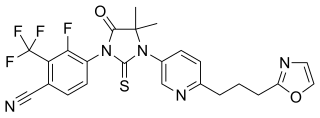
Proxalutamide is a nonsteroidal antiandrogen (NSAA) – specifically, a selective high-affinity silent antagonist of the androgen receptor (AR) – which is under development by Suzhou Kintor Pharmaceuticals, inc., a subsidiary of Kintor Pharmaceutical Limited, for the potential treatment of COVID-19, prostate cancer, and breast cancer. It was approved in Paraguay for the treatment of COVID-19 in July 2021, but has not been approved at this time in other countries.
Ketodarolutamide is a nonsteroidal antiandrogen (NSAA) and the major active metabolite of darolutamide, an NSAA which is used in the treatment of prostate cancer in men. Similarly to its parent compound, ketodarolutamide acts as a highly selective, high-affinity, competitive silent antagonist of the androgen receptor (AR). Both agents show much higher affinity and more potent inhibition of the AR relative to the other NSAAs enzalutamide and apalutamide, although they also possess much shorter and comparatively less favorable elimination half-lives. They have also been found not to activate certain mutant AR variants that enzalutamide and apalutamide do activate. Both darolutamide and ketodarolutamide show limited central nervous system distribution, indicating peripheral selectivity, and little or no inhibition or induction of cytochrome P450 enzymes such as CYP3A4, unlike enzalutamide and apalutamide.
Ralaniten is an N-terminal domain antiandrogen which was never marketed. It is a derivative of bisphenol A and one of the four stereoisomers of EPI-001. A prodrug of ralaniten, ralaniten acetate (EPI-506), was under development for the treatment of prostate cancer.

5N-Bicalutamide, or 5-azabicalutamide, is a highly potent nonsteroidal antiandrogen (NSAA) which was discovered in 2016. It is a structural modification of bicalutamide differing it from it only by the replacement of a carbon atom with a nitrogen atom in one of its phenyl rings. Similarly to bicalutamide, the drug acts as a selective antagonist of the androgen receptor (AR). However, unlike bicalutamide, it is a reversible covalent antagonist and stays bound to the receptor for a far longer amount of time. As a result of this difference, 5N-bicalutamide has markedly improved potency relative to bicalutamide, with approximately 150-fold higher affinity for the AR (Ki = 0.15 nM versus 22.3 nM) and about 20-fold greater functional inhibition (IC50Tooltip Half-maximal inhibitory concentration = 15 nM versus 310 nM) of the AR. Future studies of 5N-bicalutamide in normal and mutated prostate cancer cells are planned or underway and it is anticipated that N-bicalutamide may be able to overcome resistance. to current antiandrogens that are used in the treatment of prostate cancer.
The medical uses of bicalutamide, a nonsteroidal antiandrogen (NSAA), include the treatment of androgen-dependent conditions and hormone therapy to block the effects of androgens. Indications for bicalutamide include the treatment of prostate cancer in men, skin and hair conditions such as acne, seborrhea, hirsutism, and pattern hair loss in women, high testosterone levels in women, hormone therapy in transgender women, as a puberty blocker to prevent puberty in transgender girls and to treat early puberty in boys, and the treatment of long-lasting erections in men. It may also have some value in the treatment of paraphilias and hypersexuality in men.
Comparison of the nonsteroidal antiandrogen (NSAA) bicalutamide with other antiandrogens reveals differences between the medications in terms of efficacy, tolerability, safety, and other parameters. Relative to the other first-generation NSAAs, flutamide and nilutamide, bicalutamide shows improved potency, efficacy, tolerability, and safety, and has largely replaced these medications in clinical practice. Compared to the second-generation NSAAs, enzalutamide and apalutamide, bicalutamide has inferior potency and efficacy but similar tolerability and safety and a lower propensity for drug interactions.
The pharmacology of bicalutamide is the study of the pharmacodynamic and pharmacokinetic properties of the nonsteroidal antiandrogen (NSAA) bicalutamide. In terms of pharmacodynamics, bicalutamide acts as a selective antagonist of the androgen receptor (AR), the biological target of androgens like testosterone and dihydrotestosterone (DHT). It has no capacity to activate the AR. It does not decrease androgen levels and has no other important hormonal activity. The medication has progonadotropic effects due to its AR antagonist activity and can increase androgen, estrogen, and neurosteroid production and levels. This results in a variety of differences of bicalutamide monotherapy compared to surgical and medical castration, such as indirect estrogenic effects and associated benefits like preservation of sexual function and drawbacks like gynecomastia. Bicalutamide can paradoxically stimulate late-stage prostate cancer due to accumulated mutations in the cancer. When used as a monotherapy, bicalutamide can induce breast development in males due to its estrogenic effects. Unlike other kinds of antiandrogens, it may have less adverse effect on the testes and fertility.

RU-59063 is a nonsteroidal androgen or selective androgen receptor modulator (SARM) which was first described in 1994 and was never marketed. It was originally thought to be a potent antiandrogen, but subsequent research found that it actually possesses dose-dependent androgenic activity, albeit with lower efficacy than dihydrotestosterone (DHT). The drug is an N-substituted arylthiohydantoin and was derived from the first-generation nonsteroidal antiandrogen (NSAA) nilutamide. The second-generation NSAAs enzalutamide, RD-162, and apalutamide were derived from RU-59063.

BMS-641988 is a nonsteroidal antiandrogen which was developed by Bristol-Myers Squibb for the treatment of prostate cancer but was never marketed. It acts as a potent competitive antagonist of the androgen receptor (AR) (Ki = 10 nM; IC50Tooltip half-maximal inhibitory concentration = 56 nM). The drug was found to have 20-fold higher affinity for the AR than bicalutamide in MDA-MB-453 cells, and showed 3- to 7-fold the antiandrogenic activity of bicalutamide in vitro. It may have some weak partial agonist activity at the androgen receptor. BMS-641988 is transformed by CYP3A4 into BMS-570511, and this metabolite is then reduced to BMS-501949 by cytosolic reductases. All three compounds show similar antiandrogenic activity. In addition to its antiandrogenic activity, BMS-641988 shows activity as a negative allosteric modulator of the GABAA receptor, and can produce seizures in animals at sufficiently high doses. It also shows some drug-induced QT prolongation. BMS-641988 reached phase I clinical trials prior to the discontinuation of its development. The clinical development of BMS-641988 was terminated due to the occurrence of a seizure in a patient during a phase I study.
Masofaniten, also known by its developmental code name EPI-7386, is an N-terminal domain antiandrogen, or antagonist of the N-terminal domain (NTD) of the androgen receptor (AR), which is under development for the treatment of prostate cancer. The compound was developed as a successor of previous drugs in the EPI series such as EPI-001, ralaniten (EPI-002), and ralaniten acetate (EPI-506). Masofaniten shows 20-fold higher antiandrogenic potency than ralaniten in vitro (IC50Tooltip Half-maximal inhibitory concentration = 535 nM vs. 9,580 nM, respectively), as well as greater stability in human hepatocytes. It was planned to enter phase I clinical trials in 2020. Preliminary results of a phase I/II clinical trial were published in 2023.
