
Antiandrogens, also known as androgen antagonists or testosterone blockers, are a class of drugs that prevent androgens like testosterone and dihydrotestosterone (DHT) from mediating their biological effects in the body. They act by blocking the androgen receptor (AR) and/or inhibiting or suppressing androgen production. They can be thought of as the functional opposites of AR agonists, for instance androgens and anabolic steroids (AAS) like testosterone, DHT, and nandrolone and selective androgen receptor modulators (SARMs) like enobosarm. Antiandrogens are one of three types of sex hormone antagonists, the others being antiestrogens and antiprogestogens.

Flutamide, sold under the brand name Eulexin among others, is a nonsteroidal antiandrogen (NSAA) which is used primarily to treat prostate cancer. It is also used in the treatment of androgen-dependent conditions like acne, excessive hair growth, and high androgen levels in women. It is taken by mouth, usually three times per day.
A nonsteroidal compound is a drug that is not a steroid nor a steroid derivative. Nonsteroidal anti-inflammatory drugs (NSAIDs) are distinguished from corticosteroids as a class of anti-inflammatory agents.

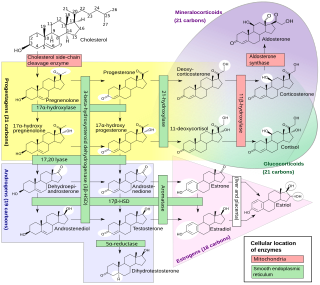
Cytochrome P450 17A1 is an enzyme of the hydroxylase type that in humans is encoded by the CYP17A1 gene on chromosome 10. It is ubiquitously expressed in many tissues and cell types, including the zona reticularis and zona fasciculata of the adrenal cortex as well as gonadal tissues. It has both 17α-hydroxylase and 17,20-lyase activities, and is a key enzyme in the steroidogenic pathway that produces progestins, mineralocorticoids, glucocorticoids, androgens, and estrogens. More specifically, the enzyme acts upon pregnenolone and progesterone to add a hydroxyl (-OH) group at carbon 17 position (C17) of the steroid D ring, or acts upon 17α-hydroxyprogesterone and 17α-hydroxypregnenolone to split the side-chain off the steroid nucleus.

Abiraterone acetate, sold under the brand name Zytiga among others, is a medication used to treat prostate cancer. Specifically it is used together with a corticosteroid for metastatic castration-resistant prostate cancer (mCRPC) and metastatic high-risk castration-sensitive prostate cancer (mCSPC). It should either be used following removal of the testicles or along with a gonadotropin-releasing hormone (GnRH) analog. It is taken by mouth.
The first antiandrogen was discovered in the 1960s. Antiandrogens antagonise the androgen receptor (AR) and thereby block the biological effects of testosterone and dihydrotestosterone (DHT). Antiandrogens are important for men with hormonally responsive diseases like prostate cancer, benign prostatic hyperplasia (BHP), acne, seborrhea, hirsutism and androgen alopecia. Antiandrogens are mainly used for the treatment of prostate diseases. Research from 2010 suggests that ARs could be linked to the disease progression of triple-negative breast cancer and salivary duct carcinoma and that antiandrogens can potentially be used to treat it.

Galeterone is a steroidal antiandrogen which was under development by Tokai Pharmaceuticals for the treatment of prostate cancer. It possesses a unique triple mechanism of action, acting as an androgen receptor antagonist, androgen receptor down regulator, and CYP17A1 inhibitor, the latter of which prevents the biosynthesis of androgens. As a CYP17A1 inhibitor, galeterone shows selectivity for 17,20-lyase over 17α-hydroxylase.
An inborn error of steroid metabolism is an inborn error of metabolism due to defects in steroid metabolism.

Oxendolone, sold under the brand names Prostetin and Roxenone, is an antiandrogen and progestin medication which is used in Japan in the treatment of enlarged prostate. However, this use is controversial due to concerns about its clinical efficacy. Oxendolone is not effective by mouth and must be given by injection into muscle.
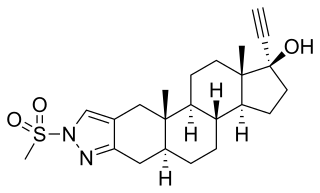
Zanoterone, also known as (5α,17α)-1'-(methylsulfonyl)-1'-H-pregn-20-yno[3,2-c]pyrazol-17-ol, is a steroidal antiandrogen which was never marketed. It was investigated for the treatment of benign prostatic hyperplasia (BPH) but failed to demonstrate sufficient efficacy in phase II clinical trials, and also showed an unacceptable incidence rate and severity of side effects. As such, it was not further developed.

A nonsteroidal estrogen is an estrogen with a nonsteroidal chemical structure. The most well-known example is the stilbestrol estrogen diethylstilbestrol (DES). Although nonsteroidal estrogens formerly had an important place in medicine, they have gradually fallen out of favor following the discovery of toxicities associated with high-dose DES starting in the early 1970s, and are now almost never used. On the other hand, virtually all selective estrogen receptor modulators (SERMs) are nonsteroidal, with triphenylethylenes like tamoxifen and clomifene having been derived from DES, and these drugs remain widely used in medicine for the treatment of breast cancer among other indications. In addition to pharmaceutical drugs, many xenoestrogens, including phytoestrogens, mycoestrogens, and synthetic endocrine disruptors like bisphenol A, are nonsteroidal substances with estrogenic activity.
A nonsteroidal antiandrogen (NSAA) is an antiandrogen with a nonsteroidal chemical structure. They are typically selective and full or silent antagonists of the androgen receptor (AR) and act by directly blocking the effects of androgens like testosterone and dihydrotestosterone (DHT). NSAAs are used in the treatment of androgen-dependent conditions in men and women. They are the converse of steroidal antiandrogens (SAAs), which are antiandrogens that are steroids and are structurally related to testosterone.

Seviteronel is an experimental cancer medication which is under development by Viamet Pharmaceuticals and Innocrin Pharmaceuticals for the treatment of prostate cancer and breast cancer. It is a nonsteroidal CYP17A1 inhibitor and works by inhibiting the production of androgens and estrogens in the body. As of July 2017, seviteronel is in phase II clinical trials for both prostate cancer and breast cancer. In January 2016, it was designated fast-track status by the United States Food and Drug Administration for prostate cancer. In April 2017, seviteronel received fast-track designation for breast cancer as well.
A steroidogenesis inhibitor, also known as a steroid biosynthesis inhibitor, is a type of drug which inhibits one or more of the enzymes that are involved in the process of steroidogenesis, the biosynthesis of endogenous steroids and steroid hormones. They may inhibit the production of cholesterol and other sterols, sex steroids such as androgens, estrogens, and progestogens, corticosteroids such as glucocorticoids and mineralocorticoids, and neurosteroids. They are used in the treatment of a variety of medical conditions that depend on endogenous steroids.

BOMT, also known by its developmental code name Ro 7-2340 and as 6α-bromo-4-oxa-17α-methyl-5α-dihydrotestosterone, is a synthetic steroidal antiandrogen which was first produced in 1970 and was never marketed for medical use. It is the 6α-brominated, 4-oxygenated, and 17α-methylated derivative of the androgen dihydrotestosterone (DHT). Along with benorterone, cyproterone, and flutamide, BOMT was among the earliest antiandrogens to be developed and extensively studied, although it is less well-documented in comparison to the others. BOMT has been investigated clinically in the treatment of benign prostatic hyperplasia, though development for this use did not continue. There was also interest in BOMT for the potential applications of acne, pattern hair loss, and possibly prostate cancer, but it was not developed for these indications either.

A steroidal antiandrogen (SAA) is an antiandrogen with a steroidal chemical structure. They are typically antagonists of the androgen receptor (AR) and act both by blocking the effects of androgens like testosterone and dihydrotestosterone (DHT) and by suppressing gonadal androgen production. SAAs lower concentrations of testosterone through simulation of the negative feedback inhibition of the hypothalamus. SAAs are used in the treatment of androgen-dependent conditions in men and women, and are also used in veterinary medicine for the same purpose. They are the converse of nonsteroidal antiandrogens (NSAAs), which are antiandrogens that are not steroids and are structurally unrelated to testosterone.
A CYP17A1 inhibitor is a type of drug which inhibits the enzyme CYP17A1. It may inhibit both of the functions of the enzyme, 17α-hydroxylase and 17,20-lyase, or may be selective for inhibition of one of these two functions. These drugs prevent the conversion of pregnane steroids into androgens like testosterone and therefore are androgen biosynthesis inhibitors and functional antiandrogens. Examples of CYP17A1 inhibitors include the older drug ketoconazole and the newer drugs abiraterone acetate, orteronel, galeterone, and seviteronel. The CYP17A1 inhibitors that have been marketed, like abiraterone acetate, are used mainly in the treatment of prostate cancer. CYP17A1 inhibitors that are not selective for inhibition of 17,20-lyase must be combined with a glucocorticoid such as prednisone in order to avoid adrenal insufficiency and mineralocorticoid excess caused by prevention of cortisol production.
An androgen synthesis inhibitor is a type of drug which inhibits the enzymatic synthesis of androgens, such as testosterone and dihydrotestosterone (DHT). They include:

7α-Thiospironolactone is a steroidal antimineralocorticoid and antiandrogen of the spirolactone group and a minor active metabolite of spironolactone. Other important metabolites of spironolactone include 7α-thiomethylspironolactone, 6β-hydroxy-7α-thiomethylspironolactone (6β-OH-7α-TMS), and canrenone (SC-9376).

The pharmacodynamics of spironolactone, an antimineralocorticoid and antiandrogen medication, concern its mechanisms of action, including its biological targets and activities, as well as its physiological effects. The pharmacodynamics of spironolactone are characterized by high antimineralocorticoid activity, moderate antiandrogenic activity, and weak steroidogenesis inhibition. In addition, spironolactone has sometimes been found to increase estradiol and cortisol levels and hence could have slight indirect estrogenic and glucocorticoid effects. The medication has also been found to interact very weakly with the estrogen and progesterone receptors, and to act as an agonist of the pregnane X receptor. Likely due to increased activation of the estrogen and/or progesterone receptors, spironolactone has very weak but significant antigonadotropic effects.
