
Levormeloxifene is a selective estrogen receptor modulator (SERM) which was being developed as an alternative to estrogen replacement therapy for the treatment and prevention of postmenopausal bone loss but did not complete development and hence was never marketed. The development was stopped because of a high incidence of gynecological side effects during clinical trials. Levormeloxifene is the levorotatory enantiomer of ormeloxifene, which, in contrast, has been marketed, though rather as a hormonal contraceptive.

Lasofoxifene, sold under the brand name Fablyn, is a nonsteroidal selective estrogen receptor modulator (SERM) which is marketed by Pfizer in Lithuania and Portugal for the prevention and treatment of osteoporosis and for the treatment of vaginal atrophy, and the result of an exclusive research collaboration with Ligand Pharmaceuticals (LGND). It also appears to have had a statistically significant effect of reducing breast cancer in women according to a study published in The Journal of the National Cancer Institute.
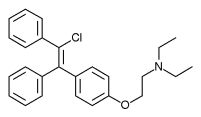
Zuclomifene (INN; or zuclomiphene (USAN)) is a nonsteroidal selective estrogen receptor modulator (SERM) of the triphenylethylene group that was never marketed. It is one of the two stereoisomers of clomifene, which itself is a mixture of 38% zuclomifene and 62% enclomifene. Zuclomifene is the (Z)-stereoisomer of clomifene, while enclomifene is the (E)-stereoisomer. Whereas zuclomifene is described as mildly estrogenic, enclomifene is described as antiestrogenic. In accordance, unlike enclomifene, zuclomifene is antigonadotropic due to activation of the estrogen receptor and reduces testosterone levels in men. It is also about five times more potent than enclomifene in inducing ovulation.

Selective androgen receptor modulators (SARMs) are a class of drugs that selectively activate the androgen receptor in specific tissues, promoting muscle and bone growth while having less effect on male reproductive tissues like the prostate gland.

Afimoxifene, also known as 4-hydroxytamoxifen (4-OHT) and by its tentative brand name TamoGel, is a selective estrogen receptor modulator (SERM) of the triphenylethylene group and an active metabolite of tamoxifen. The drug is under development under the tentative brand name TamoGel as a topical gel for the treatment of hyperplasia of the breast. It has completed a phase II clinical trial for cyclical mastalgia, but further studies are required before afimoxifene can be approved for this indication and marketed.

Arzoxifene is a selective estrogen receptor modulator (SERM) of the benzothiophene group which was never marketed. It is a potent estrogen antagonist in mammary and uterine tissue while acting as an estrogen agonist to maintain bone density and lower serum cholesterol. Arzoxifene is a highly effective agent for prevention of mammary cancer induced in the rat by the carcinogen nitrosomethylurea and is significantly more potent than raloxifene in this regard. Arzoxifene is devoid of the uterotrophic effects of tamoxifen, suggesting that, in contrast to tamoxifen, it is unlikely that the clinical use of arzoxifene will increase the risk of developing endometrial carcinoma.

Brilanestrant (INN) is a nonsteroidal combined selective estrogen receptor modulator (SERM) and selective estrogen receptor degrader (SERD) that was discovered by Aragon Pharmaceuticals and was under development by Genentech for the treatment of locally advanced or metastatic estrogen receptor (ER)-positive breast cancer.

Erteberel is a synthetic, nonsteroidal estrogen which acts as a selective ERβ agonist and was under development by Eli Lilly for the treatment of schizophrenia. It was specifically under investigation for the treatment of negative symptoms and cognitive impairment associated with the condition. It managed to reach phase II clinical trials for this indication in the United States in 2015. As of 2021 development has been discontinued. Erteberel was also under investigation for the treatment of benign prostatic hyperplasia and reached phase II clinical studies for this use but failed to improve symptoms in men with the condition and development for this indication was discontinued. The drug has also been proposed as a potential novel treatment for glioblastoma.

ICI-164384, also known as N-n-butyl-N-methyl-11-(3,17β-dihydroxyestra-1,3,5 -trien-7α-yl)undecanamide, is a steroidal antiestrogen and a synthetic derivative of estradiol which is closely related to fulvestrant and was never marketed. It is a silent antagonist of the estrogen receptor (ER) with no intrinsic estrogenic activity and hence is a pure antiestrogen, unlike selective estrogen receptor modulators (SERMs) like tamoxifen. The drug was under development by AstraZeneca for the treatment of breast cancer but was discontinued in favor of fulvestrant, which is very similar to ICI-164384 but is more potent in comparison.

Droloxifene, also known as 3-hydroxytamoxifen, is a nonsteroidal selective estrogen receptor modulator (SERM) of the triphenylethylene group that was developed originally in Germany and later in Japan for the treatment of breast cancer, osteoporosis in men and postmenopausal women, and cardiovascular disorders but was abandoned and never marketed. It reached phase II and phase III clinical trials for these indications before development was discontinued in 2000. The drug was found to be significantly less effective than tamoxifen in the treatment of breast cancer in two phase III clinical trials.

Fispemifene is a nonsteroidal selective estrogen receptor modulator (SERM) of the triphenylethylene group that was developed for the treatment of male hypogonadism but was abandoned and never marketed. It reached phase II clinical trials for this indication before development was terminated in March 2016. The drug failed to achieve statistical significance on key effectiveness endpoints in clinical trials and was discontinued by its developer for strategic reasons.

Miproxifene phosphate is a nonsteroidal selective estrogen receptor modulator (SERM) of the triphenylethylene group that was under development in Japan for the treatment of breast cancer but was abandoned and never marketed. It reached phase III clinical trials for this indication before development was discontinued. The drug is a phosphate ester and prodrug of miproxifene (DP-TAT-59) with improved water solubility that was better suited for clinical development. Miproxifene has been found to be 3- to 10-fold as potent as tamoxifen in inhibiting breast cancer cell growth in in vitro models. It is a derivative of afimoxifene (4-hydroxytamoxifen) in which an additional 4-isopropyl group is present in the β-phenyl ring.

Pipendoxifene (INN) is a nonsteroidal selective estrogen receptor modulator (SERM) that was under development by Ligand Pharmaceuticals and Wyeth-Ayerst Laboratories for the treatment of breast cancer but was not marketed. It is a member of the 2-phenylindole group of SERMs and is structurally related to zindoxifene and the marketed bazedoxifene. The drug reached phase II clinical trials before its development was discontinued. It was synthesized at the same time as bazedoxifene and was intended as a backup drug for bazedoxifene, only to be developed further if bazedoxifene had failed in clinical trials. No further development was reported after 2002 and it was formally announced that development had been terminated in November 2005.
Acolbifene/prasterone is a combination formulation of acolbifene, a selective estrogen receptor modulator, and prasterone, an androgen, estrogen, and neurosteroid, which is under development by Endoceutics for the treatment of vasomotor symptoms in postmenopausal women. It is intended for use by mouth. As of December 2017, it is in phase III clinical trials for this indication.

EM-5854 is a steroidal antiandrogen which was under development by Endoceutics, Inc. for the treatment of prostate cancer. It was first described in a patent in 2008, and was further characterized in 2012. EM-5854 reached phase I/II clinical trials for the treatment of prostate cancer but development was discontinued in March 2019.

NC 45-0095 is a synthetic nonsteroidal selective estrogen receptor modulator (SERM) which was under development by Novo Nordisk for the treatment of postmenopausal osteoporosis but was never marketed. It is a partial agonist of the estrogen receptor (IC50Tooltip half-maximal inhibitory concentration (for binding inhibition) = 9.5 nM; EC50Tooltip half-maximal effective concentration = 13 nM) with mixed estrogenic and antiestrogenic activity, and shows full estrogenic activity in bone and uterus (EmaxTooltip maximal efficacy (relative to moxestrol, in Ishikawa endometrial cancer cell line) = 105%). The compound is a pyrrolo indolizine derivative. Its development was discontinued by 2003.
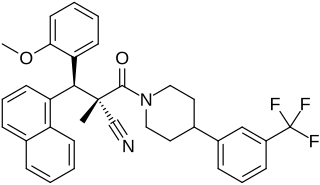
WAY-204688, also known as SIM-688, is a synthetic nonsteroidal estrogen and nuclear factor κB (NF-κB) inhibitor which was originated by ArQule and Wyeth and was under development by Wyeth for the treatment of rheumatoid arthritis, non-specific inflammation, and sepsis but was never marketed. It is a "pathway-selective" estrogen receptor (ER) ligand which inhibits NF-κB with an IC50Tooltip half-maximal inhibitory concentration of 122 nM and with maximal inhibition relative to estradiol of 94%. Inhibition of NF-κB by WAY-204688 appears to be dependent on agonism of the ERα, as it is reversed by the ERα antagonist fulvestrant, but is not dependent on the ERβ. In contrast to the case of NF-κB inhibition, WAY-204688 produces only slight elevation of creatine kinase in vitro, a measure of classical estradiol effects. It reached phase I clinical trials prior to the discontinuation of its development.
