DES was discovered in 1938 and introduced for medical use in 1939.[10][11] From about 1940 to 1971, the medication was given to pregnant women in the incorrect belief that it would reduce the risk of pregnancy complications and losses.[10] In 1971, DES was shown to cause clear-cell carcinoma, a rare vaginal tumor, in those who had been exposed to this medication in utero.[10][5] The United StatesFood and Drug Administration subsequently withdrew approval of DES as a treatment for pregnant women.[10][5] Follow-up studies have indicated that DES also has the potential to cause a variety of significant adverse medical complications during the lifetimes of those exposed including infertility.[10][12]
The United States National Cancer Institute recommends[13] children born to mothers who took DES to undergo special medical exams on a regular basis to screen for complications as a result of the medication. Individuals who were exposed to DES during their mothers' pregnancies are commonly referred to as "DES daughters" and "DES sons".[10][14] Since the discovery of the toxic effects of DES, it has largely been discontinued and is now mostly no longer marketed for human treatment.[10][15]
Interest in the use of DES to treat prostate cancer continues today.[28][29][30][31][32][33][34][excessive citations] However, use of bioidenticalparenteral estrogens like polyestradiol phosphate has been advocated in favor of oral synthetic estrogens like DES due to their much lower risk of cardiovascular toxicity.[35][32][34] In addition to prostate cancer, some interest in the use of DES to treat breast cancer continues today as well.[36][37] However, similarly to the case of prostate cancer, arguments have been made[38] for the use of bioidentical estrogens like estradiol instead of DES for breast cancer.[36][38]
In men treated with it for prostate cancer, DES has been found to produce high rates of gynecomastia (breast development) of 41 to 77%.[53]
Blood clots and cardiovascular issues
In studies of DES as a form of high-dose estrogen therapy for those with prostate cancer, it has been associated with considerable cardiovascularmorbidity and mortality.[29][5] The risk is dose-dependent.[29] A dosage of 5mg/day DES has been associated with a 36% increase in non-cancer-related (mostly cardiovascular) deaths.[29] In addition, there is an up to 15% incidence of venous thromboembolism.[54] A 3mg/day dosage of DES has been associated with an incidence of thromboembolism of 9.6 to 17%, with an incidence of cardiovascular complications of 33.3%.[29] A lower dosage of 1mg/day DES has been associated with a rate of death due to cardiovascular events of 14.8% (relative to 8.3% for orchiectomy alone).[29]
DES has been linked to a variety of long-term adverse effects in women who were treated with it during pregnancy, and/or in their offspring, including increased risk of the following:[40]
malignantuterinemesothelioma (in squirrel monkeys).[55] Evidence was also found linking ADHD to F2 generations, demonstrating that there is at least some neurological and transgenerational effects in addition to the carcinogenic.[56]
Rodent studies reveal female reproductive tract cancers and abnormalities reaching to the F2 generation, and there is evidence of adverse effects such as irregular menstrual cycles intersexual in grandchildren of DES mothers.[57] Additionally, evidence also points to transgenerational effects in F2 sons, such as hypospadias.[58] At this time however, the extent of DES transgenerational effects in humans is not fully understood.[citation needed]
Overdose
DES has been assessed in the past in clinical studies at extremely high doses of as much as 1,500 to 5,000mg/day.[36][59][60]
Pharmacology
Pharmacodynamics
Estrogenic activity
DES is an estrogen; specifically, it is a highly potent full agonist of both of the estrogen receptors (ERs).[61][62] It has approximately 468% and 295% of the affinity of estradiol at the ERα and ERβ, respectively.[63] However, EC50 values of 0.18nM and 0.06nM of DES for the ERα and ERβ, respectively, have been reported, suggesting, in spite of its binding affinity for the two receptors, several-fold preference for activation of the ERβ over the ERα.[64] In addition to the nuclear ERs, DES is an agonist of the G protein-coupled estrogen receptor (GPER), albeit with relatively low affinity (~1,000nM).[65] DES produces all of the same biological effects attributed to natural estrogens like estradiol.[66][67] This includes effects in the uterus, vagina, mammary glands, pituitary gland, and other tissues.[66][67][68][69]
A dosage of 1mg/day DES is approximately equivalent to a dosage of 50μg/day ethinylestradiol in terms of systemic estrogenic potency.[1][4] Similarly to ethinylestradiol, DES shows a marked and disproportionately strong effect on liver protein synthesis.[7] Whereas its systemic estrogenic potency was about 3.8-fold of that of estropipate (piperazine estrone sulfate), which has similar potency to micronized estradiol, the hepatic estrogenic potency of DES was 28-fold that of estropipate (or about 7.5-fold stronger potency for a dosage with equivalent systemic estrogenic effect).[1]
Notes: Values are ratios, with estradiol as standard (i.e., 1.0). Abbreviations:HF = Clinical relief of hot flashes. VE = Increased proliferation of vaginal epithelium. UCa = Decrease in UCaTooltip urinary calcium. FSH = Suppression of FSHTooltip follicle-stimulating hormone levels. LH = Suppression of LHTooltip luteinizing hormone levels. HDL-C, SHBG, CBG, and AGT = Increase in the serum levels of these liver proteins. Liver = Ratio of liver estrogenic effects to general/systemic estrogenic effects (hot flashes/gonadotropins). Sources: See template.
Testosterone levels with no treatment and with various estrogens in men with prostate cancer. Determinations were made with an early radioimmunoassay (RIA). Source was Shearer et al. (1973).Testosterone levels with placebo and 0.2 to 5mg/day diethylstilbestrol (DES) for 6months in men with prostate cancer. Determinations were made with a radioimmunoassay (RIA). Source was Kent et al. (1973).
Due to its estrogenic activity, DES has antigonadotropic effects.[83][70][94][95] That is, it exerts negative feedback on the hypothalamic–pituitary–gonadal axis (HPG axis), suppresses the secretion of the gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), and suppresses sex hormoneproduction as well as gamete production or maturation in the gonads.[83][70][94][95] A study of ovulation inhibition found that 5mg/day oral DES was 92% effective, with ovulation occurring in only a single cycle.[96][90] DES consistently suppresses testosterone levels in men into the castrate range (<50ng/dL) within 1 to 2weeks at doses of 3mg/day and above.[83][95][97] Conversely, a dosage of 1mg/day DES is unable to fully suppress testosterone levels into the castrate range in men, which instead often stabilize at just above castrate levels (>50ng/dL).[29][70][94] However, it has also been reported that 1mg/day DES results in approximately 50% suppression of testosterone levels, albeit with wide interindividual variability.[83][98] It has been said that doses of DES of less than 1mg/day have no effect on testosterone levels.[83] However, the addition of an "extremely low" dosage of 0.1mg/day DES to cyproterone acetate has been found to result in a synergistic antigonadotropic effect and to suppress testosterone levels into the castrate range in men.[99][100][101] DES at 3mg/day has similar testosterone suppression to a dose of 300mg/day, suggesting that suppression of testosterone levels is maximal by 3mg/day.[102]
Other activities
In addition to the ERs, an in vitro study found that DES also possesses activity, albeit relatively weak, at a variety of other steroid hormone receptors.[64] Whereas the study found EC50 values of 0.18nM and 0.06nM of DES for the ERα and ERβ, respectively, the medication showed significant glucocorticoid activity at a concentration of 1μM that surpassed that of 0.1nM dexamethasone, as well as significant antagonism of the androgen, progesterone, and mineralocorticoid receptors (75%, 85%, and 50% inhibition of positive control stimulation, respectively, all at a concentration of 1μM).[64] It also showed approximately 25% inhibition of the activation of PPARγ and LXRα at a concentration of 10μM.[64] The researchers stated that, to the best of their knowledge, they were the first to report such actions of DES, and hypothesized that these actions could be involved in the clinical effects of DES, for instance, in prostate cancer (notably in which particularly high dosages of DES are employed).[64] However, they also noted that the importance of the activities requires further study in animal models at pharmacologically relevant doses.[64]
DES is well-absorbed with oral administration.[1] With an oral dosage of 1mg/day DES, plasma levels of DES at 20hours following the last dose ranged between 0.9 and 1.9ng/mL (3.4 to 7.1nmol/L).[1]Sublingual administration of DES appears to have about the same estrogenic potency of oral DES in women.[105]Intrauterine DES has been studied for the treatment of uterine hypoplasia.[106] Oral DES is thought to have about 17 to 50% of the clinical estrogenic potency of DES by injection.[107]
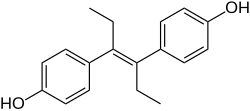
Chemical structures of estradiol and DES. Note the preservation of the two hydroxyl groups in DES and the similar distance between them relative to estradiol, which is notable when it is considered that DES was discovered serendipitously.
DES research was funded by the UK Medical Research Council (MRC), which had a policy against patenting drugs discovered using public funds. Because it was not patented, DES was produced by more than 200 pharmaceutical and chemical companies worldwide.[citation needed]
Orchiectomy or DES or both were the standard initial treatment for symptomatic advanced prostate cancer for over 40 years, until the GnRH agonistleuprorelin was found to have efficacy similar to DES without estrogenic effects and was approved in 1985.[97]
In the 1940s, DES was used off-label to prevent adverse pregnancy outcomes in women with a history of miscarriage. On July 1, 1947, the FDA approved the use of DES for this indication. The first such approval was granted to Bristol-Myers Squibb, allowing use of 25mg (and later 100mg) tablets of DES during pregnancy. Approvals were granted to other pharmaceutical companies later in the same year.[125] The recommended regimen started at 5mg per day in the seventh and eighth weeks of pregnancy (from first day of last menstrual period), increased every other week by 5mg per day through the 14th week, and then increased every week by 5mg per day from 25mg per day in the 15th week to 125mg per day in the 35th week of pregnancy.[126] DES was originally considered effective and safe for both the pregnant woman and the developing baby. It was aggressively marketed and routinely prescribed. Sales peaked in 1953.[citation needed]
In the early 1950s, a double-blind clinical trial at the University of Chicago assessed pregnancy outcomes in women who were assigned to either receive or not receive DES.[127] The study showed no benefit of taking DES during pregnancy; adverse pregnancy outcomes were not reduced in the women who were given DES. By the late 1960s, six of seven leading textbooks of obstetrics said DES was ineffective at preventing miscarriage.[125][128]
Despite an absence of evidence supporting the use of DES to prevent adverse pregnancy outcomes, DES continued to be given to pregnant women through the 1960s. In 1971, a report published in the New England Journal of Medicine showed a probable link between DES and vaginal clear cell adenocarcinoma in girls and young women who had been exposed to this drug in utero. Later in the same year, the FDA sent an FDA Drug Bulletin to all U.S. physicians advising against the use of DES in pregnant women. The FDA also removed prevention of miscarriage as an indication for DES use and added pregnancy as a contraindication for DES use.[129] On February 5, 1975, the FDA ordered 25mg and 100mg tablets of DES withdrawn, effective February 18, 1975.[130] The number of persons exposed to DES during pregnancy or in utero during the period of 1940 to 1971 is unknown, but may be as high as 2 million in the United States. DES was also used in other countries, most notably France, the Netherlands, and Great Britain.
From the 1950s through the beginning of the 1970s, DES was prescribed to prepubescent girls to begin puberty and thus stop growth by closing growth plates in the bones. Despite its clear link to cancer, doctors continued to recommend the hormone for "excess height".[131]
In 1960, DES was found to be more effective than androgens in the treatment of advanced breast cancer in postmenopausal women.[132] DES was the hormonal treatment of choice for advanced breast cancer in postmenopausal women until 1977, when the FDA approved tamoxifen, a selective estrogen receptor modulator with efficacy similar to DES but fewer side effects.[133]
In 1973, in an attempt to restrict off-label use of DES as a postcoital contraceptive (which had become prevalent at many university health services following publication of an influential study in 1971 in JAMA) to emergency situations such as rape, an FDA Drug Bulletin was sent to all U.S. physicians and pharmacists that said the FDA had approved, under restricted conditions, postcoital contraceptive use of DES.[137]
In 1975, the FDA said it had not actually given (and never did give) approval to any manufacturer to market DES as a postcoital contraceptive, but would approve that indication for emergency situations such as rape or incest if a manufacturer provided patient labeling and special packaging as set out in a FDA final rule published in 1975.[138] To discourage off-label use of DES as a postcoital contraceptive, the FDA in 1975 removed DES 25mg tablets from the market and ordered the labeling of lower doses (5mg and lower) of DES still approved for other indications changed to state: "This drug product should not be used as a postcoital contraceptive" in block capital letters on the first line of the physician prescribing information package insert and in a prominent and conspicuous location of the container and carton label.[130][139] In the 1980s, off-label use of the Yuzpe regimen of certain regular combined oral contraceptive pills superseded off-label use of DES as a postcoital contraceptive.[140]
In 1978, the FDA removed postpartum lactation suppression to prevent breast engorgement from their approved indications for DES and other estrogens.[141] In the 1990s, the only approved indications for DES were treatment of advanced prostate cancer and treatment of advanced breast cancer in postmenopausal women. The last remaining U.S. manufacturer of DES, Eli Lilly, stopped making and marketing it in 1997.[citation needed]
Trials
Diethylstilbestrol has been used countless times in studies on rats. Once it was discovered that DES was causing vaginal cancer, experiments began on both male and female rats.[142] Many of these male rats were injected with DES while other male rats were injected with olive oil, and they were considered the control group.[142] Each group received the same dosage on the same days, and the researchers performed light microscopy, electron microscopy, and confocal laser microscopy. With both the electron and confocal laser microscopy, it was prevalent that the Sertoli cells, which are somatic cells where spermatids develop in the testes, were formed 35 days later in the rats who were injected with Diethylstilbestrol compared to the rats in the control group.[142] Proceeding the completion of the trial, it was understood that rats of older age who were injected with DES experienced delay in sertoli cell maturation, underdeveloped epididymides, and drastic decrease in weight compared to its counterparts.[142]
The female rats used were inbred and most of them were given DES combined in their food. These rats were divided into three groups, one group who received no diethylstilbestrol, one group who had DES mixed into their diet, and the third group who had DES administered into their diet after day 13 of being pregnant.[143] Some rats who were given DES died before delivering their pup.[143] The group that received DES in their food for 13 days while being pregnant resulted in early abortion and delivery failure.[143] These outcomes showed that DES had a detrimental effect on pregnancy when administered as often as it was. Providing the dosing of diethylstilbestrol later in the pregnancy term also made visible the occurrence of abortions among the rats.[143] Overall, any interaction with DES in female rats concluded in the rats' experiencing abortions, improper fetal growth, and the increase in sterility.[143]
A review of people who had been treated or exposed to DES was done to find out what long-term effects would show.[144] People for a long time had been treated during their pregnancy with DES, and there have been known to be toxic and adverse effects to the hormone therapy. "Exposure to DES has been associated with an increased risk for breast cancer in DES mothers (relative risk, <2.0) and with a lifetime risk of clear-cell cervicovaginal cancer in DES daughters of 1/1000 to 1/10 000."[144] Side effects of DES are proving to be long-term as it can cause increased risks of cancer after use.[144] There will be continued work to see how far the adverse effects of DES go after previous therapy and how it will affect offspring and the mothers longer-term.[144]
Regulations
Container of "Vi-Gain" round pellets, a brand of synthetic estrogen diethylstilbestrol for veterinary use
In 1938, the ability to test the safety of DES on animals was first obtained by the FDA. The results from the preliminary tests showed that DES harmed the reproductive systems of animals. The application of these results to humans could not be determined, so the FDA could not act in a regulatory manner.[145]
New Drug Applications for DES approval were withdrawn in 1940 in a decision made by the FDA based on scientific uncertainty. However, this decision resulted in significant political pressure, so the FDA came to a compromise. The compromise meant that DES would be available only by prescription and would have to have warnings about its effects on the bottle, but the warning was dropped in 1945. In 1947, DES finally gained FDA approval for prescription to pregnant women who had diabetes as a method of preventing miscarriages. This led to the widespread prescription of DES to all pregnant women.[145]
In 1971, the FDA recommended against the prescription of DES to pregnant women.[146] As a result, DES then began to see a withdraw from the US market starting in 1972 and in the European market starting in 1978, but the FDA still did not withdraw its approval for the use of DES in humans.[147]
DES was classified as a Group 1 carcinogen by the International Agency for Research on Cancer. After classification as a carcinogen, DES had its FDA approval withdrawn in 2000.[146] DES is currently only in use for veterinary practices and in research trials as allowed by the FDA.[148]
Medical ethics
Medical Ethics in regard to the approval and use of Diethylstilbestrol have been dismissed because of the actions of the FDA and pharmaceutical companies that were making DES at the time of its use. The Vice President of the American Drug Manufacturers Association, Carson Frailey, was employed by drug companies creating DES in order to help get it approved by the Food and Drug Administration (FDA). Nancy Langston, the author of The Retreat from Precaution: Regulating Diethylstilbestrol (DES), Endocrine Disruptors, and Environmental Health, states that "Frailey persuaded fifty-four doctors from around the country to write to the FDA, describing their clinical experiences with a total of more than five thousand patients. Only four of these fifty-four doctors felt that DES should not be approved, and the result was that, against the concerns of many of the FDA medical staff, the FDA's drug chief Theodore Klumpp recommended that the FDA approve DES."[149] This excerpt describes how DES was unethically approved and shows that the motivation behind its approval was for the benefit of drug companies rather than the people who were going to use the drug. This approval of DES violates the values of medical ethics, autonomy, non-maleficence, beneficence, and justice as there was little thought put into how DES would affect its users.[150] The decisions made by the FDA leaders to approve DES without further study and convince doctors to dissimulate their opinions on the use of DES is unethical. Once DES was approved for public consumption the "warnings [for DES were] made available only on a separate circular that patients would not see. Doctors could get this warning circular only by writing to the drug companies and requesting it. Letters between companies and FDA regulators reveal that both groups feared that if a woman ever saw how many potential risks DES might present, she might refuse to take the drug—or else she might sue the company and the prescribing doctors if she did get cancer or liver damage after taking the drug."[149] Women were not informed about the possible effects of DES because doctors and FDA regulators were afraid DES would fail and never be approved costing the drug companies millions of dollars. The act of distributing potentially dangerous medicine to patients regardless of the effect and harm it may do solely for monetary gain is unethical.[citation needed]
Lawsuits
In the 1970s, the negative publicity surrounding the discovery of DES's long-term effects resulted in a huge wave of lawsuits in the United States against its manufacturers. These culminated in a landmark 1980 decision of the Supreme Court of California, Sindell v. Abbott Laboratories, in which the court imposed a rebuttable presumption of market share liability upon all DES manufacturers, proportional to their share of the market at the time the drug was consumed by the mother of a particular plaintiff.[citation needed]
Eli Lilly, a pharmaceutical company manufacturing DES, and the University of Chicago, had an action filed against them in regard to clinical trials from the 1950s. Three women filed the claim that their daughters had developments of abnormal cervical cellular formations as well as reproductive abnormalities in themselves and their sons.[151] The plaintiffs had asked the courts to certify their case as a class action but were declined by the courts. However, the courts issued an opinion that their case had merit. The court held that Eli Lilly had a duty to notify about the risks of DES once they became aware of them or should have become aware of them.[151] Under Illinois tort law, for the plaintiffs to recover under theories of breach of duty to warn and strict liability, the plaintiffs must have alleged injury to themselves. Ultimately, under their claims of breach of duty to warn and strict liability due to the plaintiffs citing risk of physical injury to others, not physical injury to themselves, the case was dismissed by the courts.[151] Although the case was not certified as class action and their claims of breach of duty to warn and strict liability was dismissed, the courts did not dismiss the battery allegations.[151] The issue was then to determine whether the University of Chicago had committed battery against these women but the case was settled before trial.[151] Part of the settlement agreement for this case, Mink v. University of Chicago, attorneys for the plaintiffs negotiated for the university to provide free medical exams for all offspring exposed to DES in utero during the 1950 experiments as well as treat the daughters of any women involved who develop DES-associated vaginal or cervical cancer.[151]
As of February 1991, there were over a thousand pending legal actions against DES manufacturers.[151] There are over 300 companies that manufactured DES according to the same formula and the largest barrier to recovery is determining which manufacturer supplied the drug in each particular case.[151] Many of the successful cases have relied on joint or several parties holding liability.
A lawsuit was filed in Boston Federal Court by 53 DES daughters who say their breast cancers were the result of DES being prescribed to their mothers while pregnant with them. Their cases survived a Daubert hearing. In 2013, the Fecho sisters who initiated the breast cancer/DES link litigation agreed to an undisclosed settlement amount on the second day of trial. The remaining litigants have received various settlements.[152]
The advocacy group DES Action USA helped provide information and support for DES-exposed persons engaged in lawsuits.[153]
Society and culture
Alan Turing, the ground-breaking cryptographer, founder of computing science and programmable computers, who also proposed the actual theoretical model of biological morphogenesis, was forcefully given this drug to induce chemical castration as a punitive and discredited "treatment" for homosexual behaviour, shortly before he died in ambiguous circumstances.[154]
At least on one occasion in New Zealand in the early 1960s, diethylstilbestrol was prescribed for the "treatment" of homosexuality.[155]
James Herriot describes a case regarding treating a small dog's testicular Sertoli cell tumor in his 1974 book All Things Bright and Beautiful. Herriot decided to prescribe a high dose of stilboestrol for the recurring tumor, with the amusing side effect that the male dog became "attractive to other male dogs", who followed the terrier around the village for a few weeks. Herriot comments in the story that he knew "The new drug was said to have a feminising effect, but surely not to that extent."[citation needed]
Veterinary use
Canine incontinence
DES has been very successful in treating female canine incontinence stemming from poor sphincter control. It is still available from compounding pharmacies, and at the low (1mg) dose, does not have the carcinogenic properties that were so problematic in humans.[156] It is generally administered once a day for seven to ten days and then once every week as needed.[citation needed]
Livestock growth promotion
The greatest usage of DES was in the livestock industry, used to improve feed conversion in beef and poultry. During the 1960s, DES was used as a growth hormone in the beef and poultry industries. It was later found to cause cancer by 1971, but was not phased out until 1979.[157][158] Although DES was discovered to be harmful to humans, its veterinary use was not immediately halted. As of 2011, DES was still being used as a growth promoter in terrestrial livestock or fish in some parts of the world including China.[159]
References
12345678910111213Chabner B, Longo DL (1996). Cancer Chemotherapy and Biotherapy: Principles and Practice. Lippincott-Raven Publishers. p.186. ISBN978-0-397-51418-2. Archived from the original on 2024-05-12. Retrieved 2020-09-03. Piperazine estrone sulfate and micronized estradiol were equipotent with respect to increases in SHBG, whereas [...] DES was 28.4-fold more potent [...]. With respect to decreased FSH, [...] DES was 3.8-fold, and ethinyl estradiol was 80 to 200-fold more potent than was piperazine estrone sulfate. The dose equivalents for ethinyl estradiol (50 μg) and DES (1 mg) reflect these relative potencies.220 [...] DES, a potent synthetic estrogen (Fig. 6-12), is absorbed well after an oral dosage. Patients given 1 mg of DES daily had plasma concentrations at 20 hours ranging from 0.9 to 1.9 ng per mL. The initial half-life of DES is 80 minutes, with a secondary half-life of 24 hours.223 The principal pathways of metabolism are conversion to the glucuronide and oxidation. The oxidative pathways include aromatic hydroxylation of the ethyl side chains and dehydrogenation to (Z,Z)-dienestrol, producing transient quinone-like intermediates that react with cellular macromolecules and cause genetic damage in eukaryotic cells.223 Metabolic activation of DES may explain its well-established carcinogenic properties.224
12345678Oelschläger H, Rothley D, Dunzendorfer U (1988). "New Results on the Pharmacokinetics of Fosfestrol". Urologia Internationalis. 43 (1): 15–23. doi:10.1159/000281427. ISSN1423-0399.
12Abramson FP, Miller HC (December 1982). "Bioavailability, distribution and pharmacokinetics of diethystilbestrol produced from stilphostrol". J Urol. 128 (6): 1336–9. doi:10.1016/s0022-5347(17)53502-8. PMID7154205.
12345678910Noller KL, Fish CR (July 1974). "Diethylstilbestrol usage: Its interesting past, important present, and questionable future". Med. Clin. North Am. 58 (4): 793–810. doi:10.1016/S0025-7125(16)32122-8. PMID4276416.
↑"DES Update: For Consumers". United States Department of Health and Human Services: Centers for Disease Control and Prevention. Archived from the original on 2020-12-11. Retrieved 2011-06-30.
↑Kreis W, Ahmann FR, Jordan VC, de Haan H, Scott M (October 1988). "Oestrogen pre-treatment abolishes luteinising hormone-releasing hormone testosterone stimulation". Br J Urol. 62 (4): 352–4. doi:10.1111/j.1464-410X.1988.tb04364.x. PMID2973364.
↑Stein BS, Smith JA (April 1985). "DES lead-in to use of luteinizing hormone releasing hormone analogs in treatment of metastatic carcinoma of prostate". Urology. 25 (4): 350–3. doi:10.1016/0090-4295(85)90484-4. PMID3920802.
↑Fernandez del Moral P, Litjens TT, Weil EH, Debruyne FM (August 1988). "Can combined DES and LHRH depot therapy (ICI 118630) prevent endocrinologic and clinical flare-up in metastatic prostate cancer?". Urology. 32 (2): 137–40. doi:10.1016/0090-4295(88)90316-0. PMID2969641.
↑Buchsbaum HJ (6 December 2012). The Menopause. Springer Science & Business Media. pp.60–. ISBN978-1-4612-5525-3. Archived from the original on 12 May 2024. Retrieved 3 September 2020.
↑Reis LO, Zani EL, García-Perdomo HA (June 2018). "Estrogen therapy in patients with prostate cancer: a contemporary systematic review". Int Urol Nephrol. 50 (6): 993–1003. doi:10.1007/s11255-018-1854-5. PMID29600433. S2CID4403709.
↑Scherr DS, Pitts WR (November 2003). "The nonsteroidal effects of diethylstilbestrol: the rationale for androgen deprivation therapy without estrogen deprivation in the treatment of prostate cancer". J. Urol. 170 (5): 1703–8. doi:10.1097/01.ju.0000077558.48257.3d. PMID14532759.
12Oh WK (September 2002). "The evolving role of estrogen therapy in prostate cancer". Clin Prostate Cancer. 1 (2): 81–9. doi:10.3816/cgc.2002.n.009. PMID15046698.
↑Malkowicz SB (August 2001). "The role of diethylstilbestrol in the treatment of prostate cancer". Urology. 58 (2 Suppl 1): 108–13. doi:10.1016/s0090-4295(01)01252-3. PMID11502463.
↑Lycette JL, Bland LB, Garzotto M, Beer TM (December 2006). "Parenteral estrogens for prostate cancer: can a new route of administration overcome old toxicities?". Clin Genitourin Cancer. 5 (3): 198–205. doi:10.3816/CGC.2006.n.037. PMID17239273.
↑Marselos M, Tomatis L (1992). "Diethylstilboestrol: I, Pharmacology, Toxicology and carcinogenicity in humans". Eur. J. Cancer. 28A (6–7): 1182–9. doi:10.1016/0959-8049(92)90482-h. PMID1627392.
12[non-primary source needed]Ellis MJ, Dehdahti F, Kommareddy A, Jamalabadi-Majidi S, Crowder R, Jeffe DB, etal. (2014). "A randomized phase 2 trial of low dose (6 mg daily) versus high dose (30 mg daily) estradiol for patients with estrogen receptor positive aromatase inhibitor resistant advanced breast cancer". Cancer Research. 69 (2 Supplement): 16. doi:10.1158/0008-5472.SABCS-16. ISSN0008-5472.
↑Hilakivi-Clarke L, de Assis S, Warri A (March 2013). "Exposures to synthetic estrogens at different times during the life, and their effect on breast cancer risk". J Mammary Gland Biol Neoplasia. 18 (1): 25–42. doi:10.1007/s10911-013-9274-8. PMC3635108. PMID23392570. From the early 1940's until 1970's, DES was given to pregnant women to prevent miscarriage, which is often proceeded by a decline in estrogen levels. It later became apparent that DES treatment was mostly ineffective in preventing miscarriage [66], but nevertheless physicians continued prescribing DES to pregnant women. A recent article summarizes the effects of maternal exposure to DES during pregnancy and its adverse effects on pregnancy and fetal development in women [67], and show that this exposure increased 2nd trimester miscarriage by 3.8 -fold.
↑Barter JF, Orr JW, Hatch KD, Shingleton HM (December 1986). "Diethylstilbestrol in pregnancy: an update". South Med J. 79 (12): 1531–4. doi:10.1097/00007611-198612000-00016. PMID3538427. S2CID33869704. Several decades ago, diethylstilbestrol (DES) was considered efficacious in improving pregnancy outcome. Later data did not support this, and the exposed mothers and offspring have suffered from a variety of problems attributed to the drug.
↑Swyer GI (April 1959). "The oestrogens". Br Med J. 1 (5128): 1029–31. doi:10.1136/bmj.1.5128.1029. PMC1993181. PMID13638626. [Diethylstilbestrol] suffers from the serious drawback that in doses above 1 mg. a day it is likely to produce nausea, vomiting, abdominal discomfort, headache, and bloating in a proportion of patients varyingly estimated from 15 to 50%.
↑Lisser H, Curtis LE (October 1947). "The syndrome of congenitally aplastic ovaries with sexual infantilism, high urinary gonadotropins, short stature and other congenital abnormalities; tabular presentation of 25 previously unpublished cases". The Journal of Clinical Endocrinology and Metabolism. 7 (10): 665–87. doi:10.1210/jcem-7-10-665. PMID20270944.
↑Di Lorenzo G, Autorino R, Perdonà S, De Placido S (December 2005). "Management of gynaecomastia in patients with prostate cancer: a systematic review". Lancet Oncol. 6 (12): 972–9. doi:10.1016/S1470-2045(05)70464-2. PMID16321765.
↑Marselos M, Tomatis L (1993). "Diethylstilboestrol: II, pharmacology, toxicology and carcinogenicity in experimental animals". European Journal of Cancer. 29A (1): 149–155. doi:10.1016/0959-8049(93)90597-9. PMID1445734.
↑Titus-Ernstoff L, Troisi R, Hatch EE, Wise LA, Palmer J, Hyer M, etal. (August 2006). "Menstrual and reproductive characteristics of women whose mothers were exposed in utero to diethylstilbestrol (DES)". International Journal of Epidemiology. 35 (4): 862–868. doi:10.1093/ije/dyl106. PMID16723367.
↑Carter AC, Sedransk N, Kelley RM, Ansfield FJ, Ravdin RG, Talley RW, Potter NR (May 1977). "Diethylstilbestrol: recommended dosages for different categories of breast cancer patients. Report of the Cooperative Breast Cancer Group". JAMA. 237 (19): 2079–8. doi:10.1001/jama.1977.03270460065023. PMID576887.
↑Karnaky KJ (December 1952). "Micronized stilbestrol for dysfunctional uterine bleeding and endometriosis". South. Med. J. 45 (12): 1166–72. doi:10.1097/00007611-195212000-00009. PMID13005120.
12Lackner JE, Tulsky AS (1941). "Effect of stilbestrol on the myometrial and endometrial activity of the human castrate uterus". The Journal of Clinical Endocrinology & Metabolism. 1 (5): 415–418. doi:10.1210/jcem-1-5-415. ISSN0021-972X. [Diethylstilbestrol], differing distinctly in chemical structure from the previously known estrogens, has been shown to produce all the biologic effects attributed to them, such as suppression of the antuitary (2), inhibition of body growth (2), proliferation of the ductile system of the breast (3), suppression of engorgement incident to lactation (4), hyperemia, edema, and distention of the uterus (5), proliferation of the endometrium (6), vaginal cornification (7), and swelling of the sexual skin (8). It likewise presumably has the supposed carcinogenic propensities of the true estrogens (9).
12Jacobsen E, Christensen SS (1939). "Comparison of the effects of stilboestrol and oestrone on the mammary tissue of castrated female rats". Acta Pathologica et Microbiologica Scandinavica. 16 (4): 359–364. doi:10.1111/j.1600-0463.1939.tb06045.x. ISSN0365-5555. After it was shown by Dodds, Goldberg, Lawson, and Robinson that stilboestrol (4.4' dioxy-α-β-diethylstilbene had the same effects as the natural oestrones on the vaginal mucosa of castrated female rats, a great number of works have appeared, which show that this substance, despite its very great chemical difference from the natural female sexual hormones has practically the same effect as these in all respects. The most important of these investigations have been made by Dodds, Lawson and Noble, by Noble, by Bishop, Boycott and Zuckermann, by Erik Guldberg, by Engelhardt, by Winterton and MacGregor, by Erik Jacobsen and most recently by Kreitmair and Sickman, by Buschbeck and Hausknecht, by Cobet, Ratschow and Stechner. The previous experiments have been made on hens, mice, rats, guineapigs, rabbits, monkeys, and human subjects.
↑Hammond CB, Maxson WS (January 1982). "Current status of estrogen therapy for the menopause". Fertility and Sterility. 37 (1): 5–25. doi:10.1016/S0015-0282(16)45970-4. PMID6277697.
↑Lauritzen C (June 1977). "[Estrogen thearpy in practice. 3. Estrogen preparations and combination preparations]" [Estrogen therapy in practice. 3. Estrogen preparations and combination preparations]. Fortschritte Der Medizin (in German). 95 (21): 1388–92. PMID559617.
↑Ryden AB (1950). "Natural and synthetic oestrogenic substances; their relative effectiveness when administered orally". Acta Endocrinologica. 4 (2): 121–39. doi:10.1530/acta.0.0040121. PMID15432047.
↑Ryden AB (1951). "The effectiveness of natural and synthetic oestrogenic substances in women". Acta Endocrinologica. 8 (2): 175–91. doi:10.1530/acta.0.0080175. PMID14902290.
↑Kottmeier HL (1947). "Ueber blutungen in der menopause: Speziell der klinischen bedeutung eines endometriums mit zeichen hormonaler beeinflussung: Part I". Acta Obstetricia et Gynecologica Scandinavica. 27 (s6): 1–121. doi:10.3109/00016344709154486. ISSN0001-6349. There is no doubt that the conversion of the endometrium with injections of both synthetic and native estrogenic hormone preparations succeeds, but the opinion whether native, orally administered preparations can produce a proliferation mucosa changes with different authors. PEDERSEN-BJERGAARD (1939) was able to show that 90% of the folliculin taken up in the blood of the vena portae is inactivated in the liver. Neither KAUFMANN (1933, 1935), RAUSCHER (1939, 1942) nor HERRNBERGER (1941) succeeded in bringing a castration endometrium into proliferation using large doses of orally administered preparations of estrone or estradiol. Other results are reported by NEUSTAEDTER (1939), LAUTERWEIN (1940) and FERIN (1941); they succeeded in converting an atrophic castration endometrium into an unambiguous proliferation mucosa with 120–300 oestradiol or with 380 oestrone.
↑Martinez-Manautou J, Rudel HW (1966). "Antiovulatory Activity of Several Synthetic and Natural Estrogens". In Robert Benjamin Greenblatt (ed.). Ovulation: Stimulation, Suppression, and Detection. Lippincott. pp.243–253.
12Herr F, Revesz C, Manson AJ, Jewell JB (1970). "Biological Properties of Estrogen Sulfates". Chemical and Biological Aspects of Steroid Conjugation. pp.368–408. doi:10.1007/978-3-642-49793-3_8. ISBN978-3-642-49506-9.
123Shearer RJ, Hendry WF, Sommerville IF, Fergusson JD (December 1973). "Plasma testosterone: an accurate monitor of hormone treatment in prostatic cancer". Br J Urol. 45 (6): 668–77. doi:10.1111/j.1464-410x.1973.tb12238.x. PMID4359746.
123Kent JR, Bischoff AJ, Arduino LJ, Mellinger GT, Byar DP, Hill M, Kozbur X (May 1973). "Estrogen dosage and suppression of testosterone levels in patients with prostatic carcinoma". J. Urol. 109 (5): 858–60. doi:10.1016/s0022-5347(17)60564-0. PMID4699685.
12The Leuprolide Study Group (November 1984). "Leuprolide versus diethylstilbestrol for metastatic prostate cancer". N Engl J Med. 311 (20): 1281–6. doi:10.1056/NEJM198411153112004. PMID6436700.
↑Goldenberg SL, Bruchovsky N, Rennie PS, Coppin CM (December 1988). "The combination of cyproterone acetate and low dose diethylstilbestrol in the treatment of advanced prostatic carcinoma". J. Urol. 140 (6): 1460–5. doi:10.1016/S0022-5347(17)42073-8. PMID2973529.
↑Goldenberg SL, Bruchovsky N, Gleave ME, Sullivan LD (June 1996). "Low-dose cyproterone acetate plus mini-dose diethylstilbestrol--a protocol for reversible medical castration". Urology. 47 (6): 882–4. doi:10.1016/S0090-4295(96)00048-9. PMID8677581.
12Ariazi EA, Jordan VC (2006). "Estrogen-related receptors as emerging targets in cancer and metabolic disorders". Curr Top Med Chem. 6 (3): 203–15. doi:10.2174/1568026610606030203. PMID16515477.
↑Friedberg V (October 1951). "Die Behandlung der genitalen Hypoplasie mit intrauterinen Cyren-B-Kristallsuspensionen" [Intrauterine Cyren-B Crystal Suspensions in Therapy of Genital Hypoplasia]. Geburtshilfe Frauenheilkd (in German). 11 (10): 923–30. ISSN0016-5751. PMID14926876.
↑Bishop PM (2008). "The Difficulty of Evaluating the Potency of Steroid Hormones by Different Routes of Administration in Humans". Ciba Foundation Symposium - Steroid Hormone Administration (Book II of Colloquia on Endocrinology, Vol. 3). Novartis Foundation Symposia. pp.349–355. doi:10.1002/9780470715154.ch10. ISBN978-0-470-71515-4. ISSN1935-4657.
↑Pugeat MM, Dunn JF, Nisula BC (July 1981). "Transport of steroid hormones: interaction of 70 drugs with testosterone-binding globulin and corticosteroid-binding globulin in human plasma". The Journal of Clinical Endocrinology and Metabolism. 53 (1): 69–75. doi:10.1210/jcem-53-1-69. PMID7195405.
↑Physicians' desk reference to pharmaceutical specialties and biologicals (15thed.). Oradell NJ: Medical Economics. 1961. p.625. ISBN0-00-093447-X.{{cite book}}: ISBN / Date incompatibility (help)
↑Dieckmann WJ, Davis ME, Rynkiewicz LM, Pottinger RE (November 1953). "Does the administration of diethylstilbestrol during pregnancy have therapeutic value?". American Journal of Obstetrics and Gynecology. 66 (5): 1062–81. doi:10.1016/S0002-9378(16)38617-3. PMID13104505.
↑United States Food and Drug Administration (1971). "Certain estrogens for oral or parenteral use. Drugs for human use; drug efficacy study implementation". Fed Regist. 36 (217): 21537–8.; 36 FR21537
12FDA (1975). "Certain estrogens for oral use. Notice of withdrawal of approval of new drug applications". Fed Regist. 40 (25): 5384.; 25 FR5384
↑Council on Drugs (1960). "Androgens and estrogens in the treatment of disseminated mammary carcinoma: retrospective study of nine hundred forty-four patients". JAMA. 172 (12): 1271–83. doi:10.1001/jama.1960.03020120049010.
↑Ingle JN, Ahmann DL, Green SJ, Edmonson JH, Bisel HF, Kvols LK, Nichols WC, Creagan ET, Hahn RG, Rubin J, Frytak S (January 1981). "Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer". The New England Journal of Medicine. 304 (1): 16–21. doi:10.1056/NEJM198101013040104. PMID7001242.
↑Goodwin WE, Cummings RH (March 1984). "Squamous metaplasia of the verumontanum with obstruction due to hypertrophy: long-term effects of estrogen on the prostate in an aging male-to-female transsexual". The Journal of Urology. 131 (3): 553–4. doi:10.1016/s0022-5347(17)50493-0. PMID6199525.
↑Seyler LE, Canalis E, Spare S, Reichlin S (July 1978). "Abnormal gonadotropin secretory responses to LRH in transsexual women after diethylstilbestrol priming". The Journal of Clinical Endocrinology and Metabolism. 47 (1): 176–83. doi:10.1210/jcem-47-1-176. PMID122396.
↑Beauchamp TL (2018). "The principles of biomedical ethics as universal principles.". Islamic Perspectives on the Principles of Biomedical Ethics: Muslim Religious Scholars and Biomedical Scientists in Face-to-Face Dialogue with Western Bioethicists. Intercultural Dialogue in Bioethics. Vol.1. pp.91–119. doi:10.1142/9781786340481_0004. ISBN978-1-78634-047-4.
12345678Mastroianni AC, Faden R, Federman D (March 1994). "Women and health research: a report from the Institute of Medicine". Kennedy Institute of Ethics Journal. 4 (1): 55–62. doi:10.1353/ken.0.0121. PMID10132589. S2CID31494934.
↑Gandhi R, Snedeker S (2000-06-01). "Consumer Concerns About Hormones in Food"(PDF). Fact Sheet #37, June 2000. Program on Breast Cancer and Environmental Risk Factors, Cornell University. Archived from the original on 2011-07-19. Retrieved 2011-07-20.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.