
Selective estrogen receptor modulators (SERMs), also known as estrogen receptor agonists/antagonists (ERAAs), are a class of drugs that act on estrogen receptors (ERs). Compared to pure ER agonists–antagonists, SERMs are more tissue-specific, allowing them to selectively inhibit or stimulate estrogen-like action in various tissues.
Tamoxifen, sold under the brand name Nolvadex among others, is a selective estrogen receptor modulator used to prevent breast cancer in women and men. It is also being studied for other types of cancer. It has been used for Albright syndrome. Tamoxifen is typically taken daily by mouth for five years for breast cancer.

Toremifene, sold under the brand name Fareston among others, is a medication which is used in the treatment of advanced breast cancer in postmenopausal women. It is taken by mouth.

Virgil Craig Jordan,, was an American and British scientist specializing in drugs for breast cancer treatment and prevention. He was Professor of Breast Medical Oncology, and Professor of Molecular and Cellular Oncology at the University of Texas MD Anderson Cancer Center, Houston, Texas. Previously, he was Scientific Director and Vice Chairman of Oncology at the Lombardi Comprehensive Cancer Center of Georgetown University. Jordan was the first to discover the breast cancer prevention properties of tamoxifen and the scientific principles for adjuvant therapy with antihormones. His later work branched out into the prevention of multiple diseases in women with the discovery of the drug group, selective estrogen receptor modulator (SERMs). He later worked on developing a new Hormone Replacement Therapy (HRT) for post-menopausal women that prevents breast cancer and does not increase the risk of breast cancer.

Chlorotrianisene (CTA), also known as tri-p-anisylchloroethylene (TACE) and sold under the brand name Tace among others, is a nonsteroidal estrogen related to diethylstilbestrol (DES) which was previously used in the treatment of menopausal symptoms and estrogen deficiency in women and prostate cancer in men, among other indications, but has since been discontinued and is now no longer available. It is taken by mouth.
Antiestrogens, also known as estrogen antagonists or estrogen blockers, are a class of drugs which prevent estrogens like estradiol from mediating their biological effects in the body. They act by blocking the estrogen receptor (ER) and/or inhibiting or suppressing estrogen production. Antiestrogens are one of three types of sex hormone antagonists, the others being antiandrogens and antiprogestogens. Antiestrogens are commonly used to stop steroid hormones, estrogen, from binding to the estrogen receptors leading to the decrease of estrogen levels. Decreased levels of estrogen can lead to complications in sexual development.

Nafoxidine or nafoxidine hydrochloride is a nonsteroidal selective estrogen receptor modulator (SERM) or partial antiestrogen of the triphenylethylene group that was developed for the treatment of advanced breast cancer by Upjohn in the 1970s but was never marketed. It was developed at around the same time as tamoxifen and clomifene, which are also triphenylethylene derivatives. The drug was originally synthesized by the fertility control program at Upjohn as a postcoital contraceptive, but was subsequently repurposed for the treatment of breast cancer. Nafoxidine was assessed in clinical trials in the treatment of breast cancer and was found to be effective. However, it produced side effects including ichthyosis, partial hair loss, and phototoxicity of the skin in almost all patients, and this resulted in the discontinuation of its development.

Trioxifene, or as the salt trioxifene mesylate (USAN), is a selective estrogen receptor modulator (SERM) with competitive binding activity against estradiol for the ERα and antagonistic activity against ERα-mediated gene expression, that was under preclinical and clinical development by Eli Lilly and Company for breast cancer and prostate cancer, but was abandoned. Its affinity for the rat estrogen receptor was reported to be 20% relative to estradiol.

Triphenylethylene (TPE) is a simple aromatic hydrocarbon that possesses weak estrogenic activity. Its estrogenic effects were discovered in 1937. TPE was derived from structural modification of the more potent estrogen diethylstilbestrol, which is a member of the stilbestrol group of nonsteroidal estrogens.

2,8-Dihydroxyhexahydrochrysene (2,8-DHHHC) is a synthetic, nonsteroidal weak estrogen with approximately 1/2,000th the estrogenic potency of the structurally-related estrogen diethylstilbestrol. It is said to be intermediate in structure between estradiol and hexestrol, but conversely to both of them, it is drastically less potent in comparison.

Ethamoxytriphetol is a synthetic nonsteroidal antiestrogen that was studied clinically in the late 1950s and early 1960s but was never marketed. MER-25 was first reported in 1958, and was the first antiestrogen to be discovered. It has been described as "essentially devoid of estrogenic activity" and as having "very low estrogenic activity in all species tested". However, some estrogenic effects in the uterus have been observed, so it is not a pure antiestrogen but is, instead, technically a selective estrogen receptor modulator (SERM). For all intents and purposes, it is a nearly pure antiestrogen, however.

Dianol is a synthetic, nonsteroidal estrogen that was never marketed. It is a dimer and impurity of anol, and was, along with hexestrol, involved in erroneous findings of highly potent estrogenic activity with anol. Although a potent estrogen, it requires a dose of 100 μg to show activity, whereas hexestrol shows activity with a mere dose of 0.2 μg.
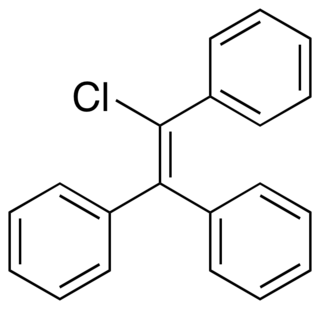
Triphenylchloroethylene, or triphenylchlorethylene, also known as chlorotriphenylethylene or as phenylstilbene chloride, is a synthetic nonsteroidal estrogen of the triphenylethylene group that was marketed in the 1940s for the treatment of menopausal symptoms, vaginal atrophy, lactation suppression, and all other estrogen-indicated conditions.

ICI-164384, also known as N-n-butyl-N-methyl-11-(3,17β-dihydroxyestra-1,3,5 -trien-7α-yl)undecanamide, is a steroidal antiestrogen and a synthetic derivative of estradiol which is closely related to fulvestrant and was never marketed. It is a silent antagonist of the estrogen receptor (ER) with no intrinsic estrogenic activity and hence is a pure antiestrogen, unlike selective estrogen receptor modulators (SERMs) like tamoxifen. The drug was under development by AstraZeneca for the treatment of breast cancer but was discontinued in favor of fulvestrant, which is very similar to ICI-164384 but is more potent in comparison.

Droloxifene, also known as 3-hydroxytamoxifen, is a nonsteroidal selective estrogen receptor modulator (SERM) of the triphenylethylene group that was developed originally in Germany and later in Japan for the treatment of breast cancer, osteoporosis in men and postmenopausal women, and cardiovascular disorders but was abandoned and never marketed. It reached phase II and phase III clinical trials for these indications before development was discontinued in 2000. The drug was found to be significantly less effective than tamoxifen in the treatment of breast cancer in two phase III clinical trials.
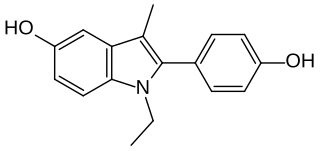
D-15414 is a nonsteroidal weak estrogen of the 2-phenylindole group which was never marketed. It is the major metabolite of the selective estrogen receptor modulator (SERM) zindoxifene (D-16726). D-15414 has high affinity for the estrogen receptor (ER) and inhibits the growth of ER-positive MCF-7 breast cancer cells in vitro. However, contradictorily, subsequent research found that the drug produced fully estrogenic effects in vitro similarly to but less actively than estradiol, with no antiestrogenic activity observed. The reason for the discrepancy between the findings is unclear, though may be due to methodology. The unexpected estrogenic activity of D-15414 may be responsible for the failure of zindoxifene in clinical trials as a treatment for breast cancer.

Panomifene is a nonsteroidal selective estrogen receptor modulator (SERM) of the triphenylethylene group related to tamoxifen that was under development as an antineoplastic agent by Egis Pharmaceuticals and IVAX Drug Research Institute in the 1990s for the treatment of breast cancer, but it was never marketed. It reached phase II clinical trials before development was terminated. The drug was described in 1981.

Nitromifene (INNTooltip International Nonproprietary Name; also as the citrate salt nitromifene citrate (USANTooltip United States Adopted Name), developmental code names CI-628, CN-5518, CN-55945) is a nonsteroidal selective estrogen receptor modulator (SERM) related to triphenylethylenes like tamoxifen that was never marketed. It is a mixture of (E)- and (Z)-isomers that possess similar antiestrogenic activity. The drug was described in 1966. Along with tamoxifen, nafoxidine, and clomifene, it was one of the earliest SERMs.
The antiestrogen withdrawal response is a paradoxical improvement in breast cancer caused by discontinuation of antiestrogen therapy for breast cancer. It has been documented rarely with the selective estrogen receptor modulators (SERMs) tamoxifen and raloxifene. The phenomenon indicates that these agents can somehow result in stimulation of breast cancer tumor progression under certain circumstances. One proposed theory for the mechanism is that the sensitivity of breast cells to estrogens shifts with estrogen deprivation, and upon antiestrogen withdrawal, endogenous estrogen acts in the manner of high-dose estrogen therapy in the breast to inhibit breast cancer growth and induce breast cancer cell death. The antiestrogen withdrawal syndrome is analogous to but less common and well-known than the antiandrogen withdrawal syndrome, a phenomenon in which paradoxical improvement in prostate cancer occurs upon discontinuation of antiandrogen therapy.
