
5α-Reductase 2 deficiency (5αR2D) is an autosomal recessive condition caused by a mutation in SRD5A2, a gene encoding the enzyme 5α-reductase type 2 (5αR2). The condition is rare, affects only genetic males, and has a broad spectrum.

Benign prostatic hyperplasia (BPH), also called prostate enlargement, is a noncancerous increase in size of the prostate gland. Symptoms may include frequent urination, trouble starting to urinate, weak stream, inability to urinate, or loss of bladder control. Complications can include urinary tract infections, bladder stones, and chronic kidney problems.
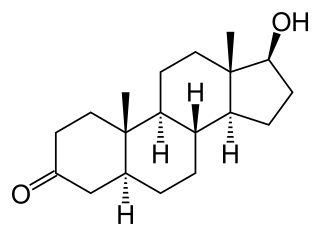
Dihydrotestosterone is an endogenous androgen sex steroid and hormone primarily involved in the growth and repair of the prostate and the penis, as well as the production of sebum and body hair composition.

Finasteride, sold under the brand names Proscar and Propecia among others, is a medication used to treat pattern hair loss and benign prostatic hyperplasia (BPH) in men. It can also be used to treat excessive hair growth in women. It is usually taken orally but there are topical formulations for patients with hair loss, designed to minimize systemic exposure by acting specifically on hair follicles.

5α-Reductases, also known as 3-oxo-5α-steroid 4-dehydrogenases, are enzymes involved in steroid metabolism. They participate in three metabolic pathways: bile acid biosynthesis, androgen and estrogen metabolism. There are three isozymes of 5α-reductase encoded by the genes SRD5A1, SRD5A2, and SRD5A3.
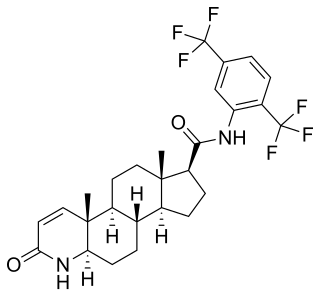
5α-Reductase inhibitors (5-ARIs), also known as dihydrotestosterone (DHT) blockers, are a class of medications with antiandrogenic effects which are used primarily in the treatment of enlarged prostate and scalp hair loss. They are also sometimes used to treat excess hair growth in women and as a component of hormone therapy for transgender women.
Dutasteride, sold under the brand name Avodart among others, is a medication primarily used to treat the symptoms of a benign prostatic hyperplasia (BPH), an enlarged prostate not associated with cancer. A few months may be required before benefits occur. It is also used for scalp hair loss in men and as a part of hormone therapy in transgender women. It is usually taken by mouth.

The human gene SRD5A2 encodes the 3-oxo-5α-steroid 4-dehydrogenase 2 enzyme, also known as 5α-reductase type 2 (5αR2), one of three isozymes of 5α-reductase.

3-Oxo-5α-steroid 4-dehydrogenase 1 is an enzyme that in humans is encoded by the SRD5A1 gene. It is one of three forms of steroid 5α-reductase.

Turosteride (FCE-26,073) is a selective inhibitor of the enzyme 5α-reductase which was under investigation by GlaxoSmithKline for the treatment of benign prostatic hyperplasia (BPH), but was never marketed. Similarly to finasteride, turosteride is selective for the type II isoform of 5α-redcutase, with about 15-fold selectivity for it over type I isoform of the enzyme. In animal studies it has been shown to inhibit prostate size and retard tumor growth. It may also be useful for the treatment of acne and hair loss.
A steroidogenesis inhibitor, also known as a steroid biosynthesis inhibitor, is a type of drug which inhibits one or more of the enzymes that are involved in the process of steroidogenesis, the biosynthesis of endogenous steroids and steroid hormones. They may inhibit the production of cholesterol and other sterols, sex steroids such as androgens, estrogens, and progestogens, corticosteroids such as glucocorticoids and mineralocorticoids, and neurosteroids. They are used in the treatment of a variety of medical conditions that depend on endogenous steroids.

BOMT, also known by its developmental code name Ro 7-2340 and as 6α-bromo-4-oxa-17α-methyl-5α-dihydrotestosterone, is a synthetic steroidal antiandrogen which was first produced in 1970 and was never marketed for medical use. It is the 6α-brominated, 4-oxygenated, and 17α-methylated derivative of the androgen dihydrotestosterone (DHT). Along with benorterone, cyproterone, and flutamide, BOMT was among the earliest antiandrogens to be developed and extensively studied, although it is less well-documented in comparison to the others. BOMT has been investigated clinically in the treatment of benign prostatic hyperplasia, though development for this use did not continue. There was also interest in BOMT for the potential applications of acne, pattern hair loss, and possibly prostate cancer, but it was not developed for these indications either.
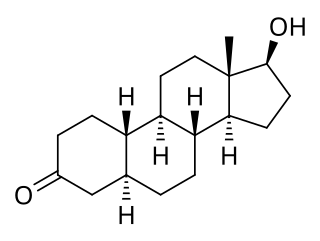
5α-Dihydronandrolone is a naturally occurring anabolic–androgenic steroid (AAS) and a 5α-reduced derivative of nandrolone (19-nortestosterone). It is a major metabolite of nandrolone and is formed from it by the actions of the enzyme 5α-reductase analogously to the formation of dihydrotestosterone (DHT) from testosterone.
An androgen synthesis inhibitor is a type of drug which inhibits the enzymatic synthesis of androgens, such as testosterone and dihydrotestosterone (DHT). They include:
Androstanolone, or stanolone, also known as dihydrotestosterone (DHT) and sold under the brand name Andractim among others, is an androgen and anabolic steroid (AAS) medication and hormone which is used mainly in the treatment of low testosterone levels in men. It is also used to treat breast development and small penis in males. Compared to testosterone, androstanolone (DHT) is less likely to aromatize into estrogen, and therefore it shows less pronounced estrogenic side effects, such as gynecomastia and water retention. On the other hand, androstanolone (DHT) show more significant androgenic side effects, such as acne, hair loss and prostate enlargement.

MK-386, also known as 4,7β-dimethyl-4-aza-5α-cholestan-3-one, is a synthetic, steroidal 5α-reductase inhibitor which was first reported in 1994 and was never marketed. It is a 4-azasteroid and a potent and selective inhibitor of 5α-reductase type I and shows high selectivity for inhibition of human 5α-reductase type I over 5α-reductase type II, with IC50 values of 0.9 nM and 154 nM, respectively. The drug was under investigation for potential treatment of androgen-dependent conditions such as acne and pattern hair loss (androgenic alopecia or baldness), but was discontinued in early clinical trials due to observations of hepatotoxicity such as elevated liver enzymes.
This article is about the discovery and development of 5α-reductase inhibitors (5-ARIs), also known as dihydrotestosterone (DHT) blockers.

MK-434 is a 5α-reductase inhibitor which was under development in the 1990s by Merck & Co for the treatment of a variety of androgen-dependent conditions including benign prostatic hyperplasia, prostate cancer, pattern hair loss, excessive hair growth, acne, and seborrhea but was never marketed. It acts as a selective inhibitor of 5α-reductase type 2. The drug has been found to decrease circulating dihydrotestosterone levels by a maximum of approximately 50% in men. MK-434 is a synthetic 4-azasteroid and is structurally related to other 5α-reductase inhibitors like finasteride.

