| |
 | |
| Clinical data | |
|---|---|
| Trade names | Erleada, others |
| Other names | ARN-509; JNJ-56021927; JNJ-927; A52 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a618018 |
| License data | |
| Pregnancy category |
|
| Routes of administration | By mouth [2] |
| Drug class | Nonsteroidal antiandrogen |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 100% [2] |
| Protein binding | Apalutamide: 96% [2] NDMA: 95% [2] |
| Metabolism | Liver (CYP2C8, CYP3A4) [2] |
| Metabolites | • NDMA [2] |
| Elimination half-life | Apalutamide: 3–4 days (at steady-state) [7] [2] |
| Excretion | Urine: 65% [2] Feces: 24% [2] |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.235.115 |
| Chemical and physical data | |
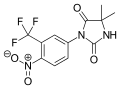
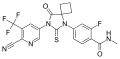
| Formula | C21H15F4N5O2S |
| Molar mass | 477.44 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
Apalutamide, sold under the brand name Erleada among others, is a nonsteroidal antiandrogen (NSAA) medication used for the treatment of prostate cancer. [2] [8] [9] It is an androgen receptor inhibitor. [2] It is taken by mouth. [2] [10]
Contents
- Medical uses
- Contraindications
- Side effects
- Overdose
- Interactions
- Pharmacology
- Pharmacodynamics
- Pharmacokinetics
- Chemistry
- History
- Society and culture
- Generic names
- Brand names
- Availability
- References
- Further reading
Side effects of apalutamide when added to castration include fatigue, nausea, abdominal pain, diarrhea, high blood pressure, rash, falls, bone fractures, and an underactive thyroid. [2] [11] [12] [10] [13] Rarely, it can cause seizures. [2] [10] The medication has a high potential for drug interactions. [2] [10] Apalutamide is an antiandrogen, and acts as an antagonist of the androgen receptor, the biological target of androgens like testosterone and dihydrotestosterone. [2] [10] [14] In doing so, it prevents the effects of these hormones in the prostate gland and elsewhere in the body. [2] [10] [14]
Apalutamide was first described in 2007, and was approved for the treatment of prostate cancer in February 2018. [8] [9] [10] [15] It is the first medication to be approved specifically for the treatment of non-metastatic castration-resistant prostate cancer. [2] [10] [9]