GABA is the chief inhibitory neurotransmitter in the developmentally mature mammalian central nervous system. Its principal role is reducing neuronal excitability throughout the nervous system.

Nootropics, colloquially brain supplements, smart drugs and cognitive enhancers, are natural, semisynthetic or synthetic compounds which purportedly improve cognitive functions, such as executive functions, attention or memory.

Piracetam is a drug that has efficacy in cognitive disorders, vertigo, cortical myoclonus, dyslexia, and sickle cell anemia; sources differ on its usefulness for dementia. Piracetam is sold as a medication in many European countries. Sale of piracetam is not illegal in the United States, although it is not regulated nor approved by the FDA, so it is legally sold for research use only.

The GABA receptors are a class of receptors that respond to the neurotransmitter gamma-aminobutyric acid (GABA), the chief inhibitory compound in the mature vertebrate central nervous system. There are two classes of GABA receptors: GABAA and GABAB. GABAA receptors are ligand-gated ion channels ; whereas GABAB receptors are G protein-coupled receptors, also called metabotropic receptors.

Ibotenic acid or (S)-2-amino-2-(3-hydroxyisoxazol-5-yl)acetic acid, also referred to as ibotenate, is a chemical compound and psychoactive drug which occurs naturally in Amanita muscaria and related species of mushrooms typically found in the temperate and boreal regions of the northern hemisphere. It is a prodrug of muscimol, broken down by the liver to that much more stable compound. It is a conformationally-restricted analogue of the neurotransmitter glutamate, and due to its structural similarity to this neurotransmitter, acts as a non-selective glutamate receptor agonist. Because of this, ibotenic acid can be a powerful neurotoxin in high doses, and is employed as a "brain-lesioning agent" through cranial injections in scientific research. The neurotoxic effects appear to be dose-related and risks are unclear through consumption of ibotenic-acid containing fungi, although thought to be negligible in small doses.

Muscimol is one of the principal psychoactive constituents of Amanita muscaria and related species of mushroom. Muscimol is a potent and selective orthosteric agonist for the GABAA receptor and displays sedative-hypnotic, depressant and hallucinogenic psychoactivity. This colorless or white solid is classified as an isoxazole.

Aniracetam, also known as N-anisoyl-2-pyrrolidinone, is a racetam which is sold in Europe as a prescription drug. It is not approved by the Food and Drug Administration for use in the United States as a prescription medication or dietary supplement. Despite the FDA's lack of approval, the drug is readily available over-the-counter in misbranded dietary supplements.

Oxiracetam is a nootropic drug of the racetam family and a very mild stimulant. Several studies suggest that the substance is safe even when high doses are consumed for a long period of time. However, the mechanism of action of the racetam drug family is still a matter of research. Oxiracetam is not approved by Food and Drug Administration for any medical use in the United States.

Nebracetam is an investigational drug of the racetam family that is a M1 acetylcholine receptor agonist in rats. Based on a human leukemic T cell experiment in 1991, it is believed to act as an agonist for human M1-muscarinic receptors. It is also believed to act as a nootropic, like many other racetam drugs. A chemoenzymatic method of synthesis was reported in 2008. As of 2023, human trials have not yet been conducted.

The GABAA receptor (GABAAR) is an ionotropic receptor and ligand-gated ion channel. Its endogenous ligand is γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the central nervous system. Accurate regulation of GABAergic transmission through appropriate developmental processes, specificity to neural cell types, and responsiveness to activity is crucial for the proper functioning of nearly all aspects of the central nervous system (CNS). Upon opening, the GABAA receptor on the postsynaptic cell is selectively permeable to chloride ions and, to a lesser extent, bicarbonate ions.
Racetams are a class of drugs that share a pyrrolidone nucleus. Some, such as piracetam, aniracetam, oxiracetam, pramiracetam and phenylpiracetam are considered nootropics. Others such as levetiracetam, brivaracetam, and seletracetam are anticonvulsants.
The GABAA-rho receptor is a subclass of GABAA receptors composed entirely of rho (ρ) subunits. GABAA receptors including those of the ρ-subclass are ligand-gated ion channels responsible for mediating the effects of gamma-amino butyric acid (GABA), the major inhibitory neurotransmitter in the brain. The GABAA-ρ receptor, like other GABAA receptors, is expressed in many areas of the brain, but in contrast to other GABAA receptors, the GABAA-ρ receptor has especially high expression in the retina.
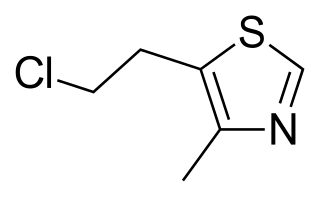
Clomethiazole is a sedative and hypnotic originally developed by Hoffmann-La Roche in the 1930s. The drug is used in treating and preventing symptoms of acute alcohol withdrawal.

Tetramethylenedisulfotetramine (TETS) is an organic compound used as a rodenticide. It is an odorless, tasteless white powder that is slightly soluble in water, DMSO and acetone, and insoluble in methanol and ethanol. It is a sulfamide derivative. It can be synthesized by reacting sulfamide with formaldehyde solution in acidified water. When crystallized from acetone, it forms cubic crystals with a melting point of 255–260 °C.
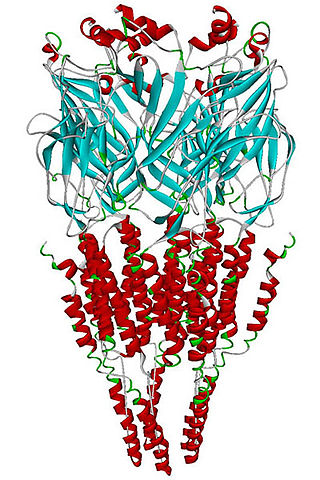
The alpha-7 nicotinic receptor, also known as the α7 receptor, is a type of nicotinic acetylcholine receptor implicated in long-term memory, consisting entirely of α7 subunits. As with other nicotinic acetylcholine receptors, functional α7 receptors are pentameric [i.e., (α7)5 stoichiometry].

L-655,708 (FG-8094) is a nootropic drug invented in 1996 by a team working for Merck, Sharp and Dohme, that was the first compound developed which acts as a subtype-selective inverse agonist at the α5 subtype of the benzodiazepine binding site on the GABAA receptor. It acts as an inverse agonist at the α1, α2, α3 and α5 subtypes, but with much higher affinity for α5, and unlike newer α5 inverse agonists such as α5IA, L-655,708 exerts its subtype selectivity purely via higher binding affinity for this receptor subtype, with its efficacy as an inverse agonist being around the same at all the subtypes it binds to.
A convulsant is a drug which induces convulsions and/or epileptic seizures, the opposite of an anticonvulsant. These drugs generally act as stimulants at low doses, but are not used for this purpose due to the risk of convulsions and consequent excitotoxicity. Most convulsants are antagonists at either the GABAA or glycine receptors, or ionotropic glutamate receptor agonists. Many other drugs may cause convulsions as a side effect at high doses but only drugs whose primary action is to cause convulsions are known as convulsants. Nerve agents such as sarin, which were developed as chemical weapons, produce convulsions as a major part of their toxidrome, but also produce a number of other effects in the body and are usually classified separately. Dieldrin which was developed as an insecticide blocks chloride influx into the neurons causing hyperexcitability of the CNS and convulsions. The Irwin observation test and other studies that record clinical signs are used to test the potential for a drug to induce convulsions. Camphor, and other terpenes given to children with colds can act as convulsants in children who have had febrile seizures.

SH-053-R-CH3-2′F is a drug used in scientific research which is a benzodiazepine derivative. It produces some of the same effects as other benzodiazepines, but is much more subtype-selective than most other drugs of this class, having high selectivity, binding affinity and efficacy at the α5 subtype of the GABAA receptor. This gives much tighter control of the effects produced, and so while SH-053-R-CH3-2′F retains sedative and anxiolytic effects, it does not cause ataxia at moderate doses. SH-053-R-CH3-2′F also blocks the nootropic effects of the α5-selective inverse agonist PWZ-029, so amnesia is also a likely side effect.

Quisqualamine is the α-decarboxylated analogue of quisqualic acid, as well as a relative of the neurotransmitters glutamate and γ-aminobutyric acid (GABA). α-Decarboxylation of excitatory amino acids can produce derivatives with inhibitory effects. Indeed, unlike quisqualic acid, quisqualamine has central depressant and neuroprotective properties and appears to act predominantly as an agonist of the GABAA receptor and also to a lesser extent as an agonist of the glycine receptor, due to the facts that its actions are inhibited in vitro by GABAA antagonists like bicuculline and picrotoxin and by the glycine antagonist strychnine, respectively. Mg2+ and DL-AP5, NMDA receptor blockers, CNQX, an antagonist of both the AMPA and kainate receptors, and 2-hydroxysaclofen, a GABAB receptor antagonist, do not affect quisqualamine's actions in vitro, suggesting that it does not directly affect the ionotropic glutamate receptors or the GABAB receptor in any way. Whether it binds to and acts upon any of the metabotropic glutamate receptors like its analogue quisqualic acid however is unclear.