In biochemistry and pharmacology, receptors are chemical structures, composed of protein, that receive and transduce signals that may be integrated into biological systems. These signals are typically chemical messengers, which bind to a receptor, they cause some form of cellular/tissue response, e.g. a change in the electrical activity of a cell. There are three main ways the action of the receptor can be classified: relay of signal, amplification, or integration. Relaying sends the signal onward, amplification increases the effect of a single ligand, and integration allows the signal to be incorporated into another biochemical pathway. In this sense, a receptor is a protein-molecule that recognizes and responds to endogenous chemical signals. For example, an acetylcholine receptor recognizes and responds to its endogenous ligand, acetylcholine. However, sometimes in pharmacology, the term is also used to include other proteins that are drug targets, such as enzymes, transporters, and ion channels.

A receptor antagonist is a type of receptor ligand or drug that blocks or dampens a biological response by binding to and blocking a receptor rather than activating it like an agonist. They are sometimes called blockers; examples include alpha blockers, beta blockers, and calcium channel blockers. In pharmacology, antagonists have affinity but no efficacy for their cognate receptors, and binding will disrupt the interaction and inhibit the function of an agonist or inverse agonist at receptors. Antagonists mediate their effects by binding to the active site or to the allosteric site on a receptor, or they may interact at unique binding sites not normally involved in the biological regulation of the receptor's activity. Antagonist activity may be reversible or irreversible depending on the longevity of the antagonist–receptor complex, which, in turn, depends on the nature of antagonist–receptor binding. The majority of drug antagonists achieve their potency by competing with endogenous ligands or substrates at structurally defined binding sites on receptors.

Ligand-gated ion channels (LICs, LGIC), also commonly referred to as ionotropic receptors, are a group of transmembrane ion-channel proteins which open to allow ions such as Na+, K+, Ca2+, and/or Cl− to pass through the membrane in response to the binding of a chemical messenger (i.e. a ligand), such as a neurotransmitter.

The GABAA receptor (GABAAR) is an ionotropic receptor and ligand-gated ion channel. Its endogenous ligand is γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the central nervous system. Upon activation, the GABAA receptor selectively conducts Cl− through its pore. Cl- will flow out of the cell if the internal voltage is less than resting potential and Cl- will flow in if it is more than resting potential. This causes an inhibitory effect on neurotransmission by diminishing the chance of a successful action potential occurring. The reversal potential of the GABAA-mediated inhibitory postsynaptic potential (IPSP) in normal solution is −70 mV, contrasting the GABAB IPSP (-100 mV).

Bretazenil (Ro16-6028) is an imidazopyrrolobenzodiazepine anxiolytic drug which is derived from the benzodiazepine family, and was invented in 1988. It is most closely related in structure to the GABA antagonist flumazenil, although its effects are somewhat different. It is classified as a high-potency benzodiazepine due to its high affinity binding to benzodiazepine binding sites where it acts as a partial agonist. Its profile as a partial agonist and preclinical trial data suggests that it may have a reduced adverse effect profile. In particular bretazenil has been proposed to cause a less strong development of tolerance and withdrawal syndrome. Bretazenil differs from traditional 1,4-benzodiazepines by being a partial agonist and because it binds to α1, α2, α3, α4, α5 and α6 subunit containing GABAA receptor benzodiazepine receptor complexes. 1,4-benzodiazepines bind only to α1, α2, α3 and α5GABAA benzodiazepine receptor complexes.

DMCM (methyl-6,7-dimethoxy-4-ethyl-beta-carboline-3-carboxylate) is a drug from the beta-carboline family. It acts as a negative allosteric modulator of GABAA receptors, meaning that it causes the opposite effects to the benzodiazepine class of drugs. As such, DMCM has anxiogenic and convulsant properties, and is used in scientific research to induce anxiety so that new anxiolytic medications can be tested, and to produce convulsions so that anticonvulsant medications can be tested. It has also been shown to produce analgesic effects in animals, thought to be because it produces panic which reduces the perception of pain.
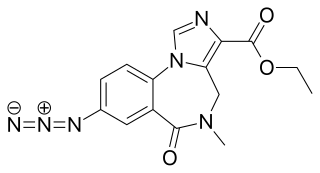
Ro15-4513 is a weak partial inverse agonist of the benzodiazepine class of drugs, developed by Hoffmann–La Roche in the 1980s. It acts as a competitive antagonist, and can therefore be an antidote to the acute impairment caused by alcohols, including ethanol, isopropanol, tert-butyl alcohol, tert-butyl alcohol, tert-amyl alcohol, 3-methyl-3-pentanol, methylpentynol and ethchlorvynol.

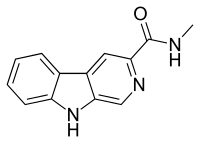
Imidazenil is an experimental anxiolytic drug which is derived from the benzodiazepine family, and is most closely related to other imidazobenzodiazepines such as midazolam, flumazenil, and bretazenil.

Loreclezole is a sedative and an anticonvulsant which acts as a GABAA receptor positive allosteric modulator. The binding site of loreclezole has been shown experimentally to be shared by valerenic acid, an extract of the root of the valerian plant. Structurally, loreclezole is a triazole derivative. In animal seizure models, loreclezole is protective against pentylenetetrazol seizures but is less active in the maximal electroshock test. In addition, at low, nontoxic doses, the drug has anti-absence activity in a genetic model of generalized absence epilepsy. Consequently, loreclezole has a profile of activity similar to that of benzodiazepines. A potential benzodiazepine-like interaction with GABA receptors is suggested by the observation that the anticonvulsant effects of loreclezole can be reversed by benzodiazepine receptor inverse agonists. The benzodiazepine antagonist flumazenil, however, fails to alter the anticonvulsant activity of loreclezole, indicating that loreclezole is not a benzodiazepine receptor agonist. Using native rat and cloned human GABA-A receptors, loreclezole strongly potentiated GABA-activated chloride current. However, activity of the drug did not require the presence of the γ-subunit and was not blocked by flumazenil, confirming that loreclezole does not interact with the benzodiazepine recognition site.

The alpha-7 nicotinic receptor, also known as the α7 receptor, is a type of nicotinic acetylcholine receptor implicated in long term memory, consisting entirely of α7 subunits. As with other nicotinic acetylcholine receptors, functional α7 receptors are pentameric [i.e., (α7)5 stoichiometry].

TPA-023 (MK-0777) is an anxiolytic drug with a novel chemical structure, which is used in scientific research. It has similar effects to benzodiazepine drugs, but is structurally distinct and so is classed as a nonbenzodiazepine anxiolytic. It is a subtype-selective, mixed agonist-antagonist at GABAA receptors, which acts as a partial agonist at the α2 and α3 subtypes, but as a silent antagonist at α1 and α5 subtypes. It has primarily anxiolytic and anticonvulsant effects in animal tests, but with no sedative effects even at 50 times the effective anxiolytic dose.

α5IA (LS-193,268) is a nootropic drug invented in 2004 by a team working for Merck, Sharp and Dohme, which acts as a subtype-selective inverse agonist at the benzodiazepine binding site on the GABAA receptor. It binds to the α1, α2, α3 and α5 subtypes, but shows much higher efficacy at the α5 subtype, and acts either as a weak partial agonist or inverse agonist at the other subtypes, with its partial agonist effect at α2 likely to be responsible for the lack of anxiety produced by this drug when compared to older α5-preferring inverse agonists such as L-655,708.

L-655,708 (FG-8094) is a nootropic drug invented in 1996 by a team working for Merck, Sharp and Dohme, that was the first compound developed which acts as a subtype-selective inverse agonist at the α5 subtype of the benzodiazepine binding site on the GABAA receptor. It acts as an inverse agonist at the α1, α2, α3 and α5 subtypes, but with much higher affinity for α5, and unlike newer α5 inverse agonists such as α5IA, L-655,708 exerts its subtype selectivity purely via higher binding affinity for this receptor subtype, with its efficacy as an inverse agonist being around the same at all the subtypes it binds to.
A convulsant is a drug which induces convulsions and/or epileptic seizures, the opposite of an anticonvulsant. These drugs generally act as stimulants at low doses, but are not used for this purpose due to the risk of convulsions and consequent excitotoxicity. Most convulsants are antagonists at either the GABAA or glycine receptors, or ionotropic glutamate receptor agonists. Many other drugs may cause convulsions as a side effect at high doses but only drugs whose primary action is to cause convulsions are known as convulsants. Nerve agents such as sarin, which were developed as chemical weapons, produce convulsions as a major part of their toxidrome, but also produce a number of other effects in the body and are usually classified separately.

ZK-93426 (ethyl-5-isopropoxy-4-methyl-beta-carboline-3-carboxylate) is a drug from the beta-carboline family. It acts as a weak partial inverse agonist of benzodiazepine receptors, meaning that it causes the opposite effects to the benzodiazepine class of drugs and has anxiogenic properties, although unlike most benzodiazepine antagonists it is not a convulsant and actually has weak anticonvulsant effects. In human tests it produced alertness, restlessness and feelings of apprehension, and reversed the effect of the benzodiazepine lormetazepam. It was also shown to produce nootropic effects and increased release of acetylcholine.

PWZ-029 is a benzodiazepine derivative drug with nootropic effects developed by WiSys, It acts as a subtype-selective, mixed agonist-inverse agonist at the benzodiazepine binding site on the GABAA receptor, acting as a partial inverse agonist at the α5 subtype and a weak partial agonist at the α3 subtype. This gives it a mixed pharmacological profile, producing at low doses memory-enhancing effects but with no convulsant or anxiogenic effects or muscle weakness, although at higher doses it produces some sedative effects.
In pharmacology and biochemistry, allosteric modulators are a group of substances that bind to a receptor to change that receptor's response to stimulus. Some of them, like benzodiazepines, are drugs. The site that an allosteric modulator binds to is not the same one to which an endogenous agonist of the receptor would bind. Modulators and agonists can both be called receptor ligands.

FG-8205 (L-663,581) is an imidazobenzodiazepine derivative related to bretazenil, which acts as a partial agonist at GABAA receptors, with slight selectivity for the α1-containing subtype. In animal tests it has anxiolytic and anticonvulsant effects but with little sedation or ataxia produced.

Iomazenil is an antagonist and partial inverse agonist of benzodiazepine and a potential treatment for alcohol abuse. The compound was introduced in 1989 by pharmaceutical company Hoffmann-La Roche as an Iodine-123-labelled SPECT tracer for imaging benzodiazepine receptors in the brain. Iomazenil is an analogue of flumazenil (Ro15-1788).
A GABAA receptor negative allosteric modulator is a negative allosteric modulator (NAM), or inhibitor, of the GABAA receptor, a ligand-gated ion channel of the major inhibitory neurotransmitter γ-aminobutyric acid (GABA). They are closely related and similar to GABAA receptor antagonists. The effects of GABAA receptor NAMs are functionally the opposite of those of GABAA receptor positive allosteric modulators (PAMs) like the benzodiazepines, barbiturates, and ethanol (alcohol). Non-selective GABAA receptor NAMs can produce a variety of effects including convulsions, neurotoxicity, and anxiety, among others.