Related Research Articles

In chemistry, a reagent or analytical reagent is a substance or compound added to a system to cause a chemical reaction, or test if one occurs. The terms reactant and reagent are often used interchangeably, but reactant specifies a substance consumed in the course of a chemical reaction. Solvents, though involved in the reaction mechanism, are usually not called reactants. Similarly, catalysts are not consumed by the reaction, so they are not reactants. In biochemistry, especially in connection with enzyme-catalyzed reactions, the reactants are commonly called substrates.
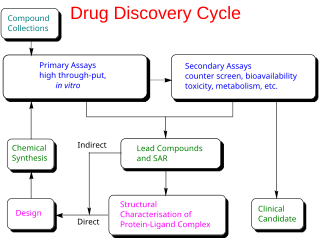
In the fields of medicine, biotechnology, and pharmacology, drug discovery is the process by which new candidate medications are discovered.
Drug design, often referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques. This type of modeling is sometimes referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design. In addition to small molecules, biopharmaceuticals including peptides and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also been developed.

High-throughput screening (HTS) is a method for scientific discovery especially used in drug discovery and relevant to the fields of biology, materials science and chemistry. Using robotics, data processing/control software, liquid handling devices, and sensitive detectors, high-throughput screening allows a researcher to quickly conduct millions of chemical, genetic, or pharmacological tests. Through this process one can quickly recognize active compounds, antibodies, or genes that modulate a particular biomolecular pathway. The results of these experiments provide starting points for drug design and for understanding the noninteraction or role of a particular location.

Medicinal or pharmaceutical chemistry is a scientific discipline at the intersection of chemistry and pharmacy involved with designing and developing pharmaceutical drugs. Medicinal chemistry involves the identification, synthesis and development of new chemical entities suitable for therapeutic use. It also includes the study of existing drugs, their biological properties, and their quantitative structure-activity relationships (QSAR).
Chemogenomics, or chemical genomics, is the systematic screening of targeted chemical libraries of small molecules against individual drug target families with the ultimate goal of identification of novel drugs and drug targets. Typically some members of a target library have been well characterized where both the function has been determined and compounds that modulate the function of those targets have been identified. Other members of the target family may have unknown function with no known ligands and hence are classified as orphan receptors. By identifying screening hits that modulate the activity of the less well characterized members of the target family, the function of these novel targets can be elucidated. Furthermore, the hits for these targets can be used as a starting point for drug discovery. The completion of the human genome project has provided an abundance of potential targets for therapeutic intervention. Chemogenomics strives to study the intersection of all possible drugs on all of these potential targets.
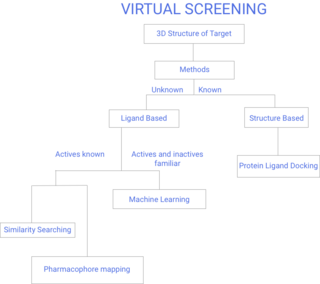
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.
High-content screening (HCS), also known as high-content analysis (HCA) or cellomics, is a method that is used in biological research and drug discovery to identify substances such as small molecules, peptides, or RNAi that alter the phenotype of a cell in a desired manner. Hence high content screening is a type of phenotypic screen conducted in cells involving the analysis of whole cells or components of cells with simultaneous readout of several parameters. HCS is related to high-throughput screening (HTS), in which thousands of compounds are tested in parallel for their activity in one or more biological assays, but involves assays of more complex cellular phenotypes as outputs. Phenotypic changes may include increases or decreases in the production of cellular products such as proteins and/or changes in the morphology of the cell. Hence HCA typically involves automated microscopy and image analysis. Unlike high-content analysis, high-content screening implies a level of throughput which is why the term "screening" differentiates HCS from HCA, which may be high in content but low in throughput.
Compound management in the field of drug discovery refers to the systematic collection, storage, retrieval, and quality control of small molecule chemical compounds used in high-throughput screening and other research activities to identify hits that can be developed into candidate drugs.
A chemical library or compound library is a collection of stored chemicals usually used ultimately in high-throughput screening or industrial manufacture. The chemical library can consist in simple terms of a series of stored chemicals. Each chemical has associated information stored in some kind of database with information such as the chemical structure, purity, quantity, and physiochemical characteristics of the compound.
A lead compound in drug discovery is a chemical compound that has pharmacological or biological activity likely to be therapeutically useful, but may nevertheless have suboptimal structure that requires modification to fit better to the target; lead drugs offer the prospect of being followed by back-up compounds. Its chemical structure serves as a starting point for chemical modifications in order to improve potency, selectivity, or pharmacokinetic parameters. Furthermore, newly invented pharmacologically active moieties may have poor druglikeness and may require chemical modification to become drug-like enough to be tested biologically or clinically.
High throughput biology is the use of automation equipment with classical cell biology techniques to address biological questions that are otherwise unattainable using conventional methods. It may incorporate techniques from optics, chemistry, biology or image analysis to permit rapid, highly parallel research into how cells function, interact with each other and how pathogens exploit them in disease.
Fragment-based lead discovery (FBLD) also known as fragment-based drug discovery (FBDD) is a method used for finding lead compounds as part of the drug discovery process. Fragments are small organic molecules which are small in size and low in molecular weight. It is based on identifying small chemical fragments, which may bind only weakly to the biological target, and then growing them or combining them to produce a lead with a higher affinity. FBLD can be compared with high-throughput screening (HTS). In HTS, libraries with up to millions of compounds, with molecular weights of around 500 Da, are screened, and nanomolar binding affinities are sought. In contrast, in the early phase of FBLD, libraries with a few thousand compounds with molecular weights of around 200 Da may be screened, and millimolar affinities can be considered useful. FBLD is a technique being used in research for discovering novel potent inhibitors. This methodology could help to design multitarget drugs for multiple diseases. The multitarget inhibitor approach is based on designing an inhibitor for the multiple targets. This type of drug design opens up new polypharmacological avenues for discovering innovative and effective therapies. Neurodegenerative diseases like Alzheimer’s (AD) and Parkinson’s, among others, also show rather complex etiopathologies. Multitarget inhibitors are more appropriate for addressing the complexity of AD and may provide new drugs for controlling the multifactorial nature of AD, stopping its progression.
Dipeptidyl peptidase-4 inhibitors are enzyme inhibitors that inhibit the enzyme dipeptidyl peptidase-4 (DPP-4). They are used in the treatment of type 2 diabetes mellitus. Inhibition of the DPP-4 enzyme prolongs and enhances the activity of incretins that play an important role in insulin secretion and blood glucose control regulation. Type 2 diabetes mellitus is a chronic metabolic disease that results from inability of the β-cells in the pancreas to secrete sufficient amounts of insulin to meet the body's needs. Insulin resistance and increased hepatic glucose production can also play a role by increasing the body's demand for insulin. Current treatments, other than insulin supplementation, are sometimes not sufficient to achieve control and may cause undesirable side effects, such as weight gain and hypoglycemia. In recent years, new drugs have been developed, based on continuing research into the mechanism of insulin production and regulation of the metabolism of sugar in the body. The enzyme DPP-4 has been found to play a significant role.
Ligand efficiency is a measurement of the binding energy per atom of a ligand to its binding partner, such as a receptor or enzyme.
A thermal shift assay (TSA) measures changes in the thermal denaturation temperature and hence stability of a protein under varying conditions such as variations in drug concentration, buffer formulation, redox potential, or sequence mutation. The most common method for measuring protein thermal shifts is differential scanning fluorimetry (DSF). DSF methodology includes techniques such as nanoDSF, which relies on the intrinsic fluorescence from native tryptophan or tyrosine residues, and Thermofluor, which utilizes extrinsic fluorogenic dyes.
Chemoproteomics entails a broad array of techniques used to identify and interrogate protein-small molecule interactions. Chemoproteomics complements phenotypic drug discovery, a paradigm that aims to discover lead compounds on the basis of alleviating a disease phenotype, as opposed to target-based drug discovery, in which lead compounds are designed to interact with predetermined disease-driving biological targets. As phenotypic drug discovery assays do not provide confirmation of a compound's mechanism of action, chemoproteomics provides valuable follow-up strategies to narrow down potential targets and eventually validate a molecule's mechanism of action. Chemoproteomics also attempts to address the inherent challenge of drug promiscuity in small molecule drug discovery by analyzing protein-small molecule interactions on a proteome-wide scale. A major goal of chemoproteomics is to characterize the interactome of drug candidates to gain insight into mechanisms of off-target toxicity and polypharmacology.

James Inglese is an American biochemist, the director of the Assay Development and Screening Technology Laboratory at the National Center for Advancing Translational Sciences, a Center within the National Institutes of Health. His specialty is small molecule high throughput screening. Inglese's laboratory develops methods and strategies in molecular pharmacology with drug discovery applications. The work of his research group and collaborators focuses on genetic and infectious disease-associated biology.
Jonathan Baell is trained as an Australian medicinal chemist and is currently executive director, early leads chemistry at Lyterian Therapeutics in San Francisco. Prior to this, he was a research professor in medicinal chemistry at the Monash Institute of Pharmaceutical Sciences (MIPS), the director of the Australian Translational Medicinal Chemistry Facility and a Chief Investigator at the ARC Centre for Fragment-Based Design. He was President of the International Chemical Biology Society 2018-2021 and is currently chair of the board. His research focuses on the early stages of drug discovery, including high-throughput screening (HTS) library design, hit-to-lead and lead optimization for the treatment of a variety of diseases, such as malaria and neglected diseases.
Klara Valko is a scientist, consultant, academic and author. She is the director of Bio-Mimetic Chromatography as well as an honorary professor at University College London School of Pharmacy.
References
- ↑ Deprez-Poulain R, Deprez B (2004). "Facts, figures and trends in lead generation". Current Topics in Medicinal Chemistry. 4 (6): 569–80. doi:10.2174/1568026043451168. PMID 14965294.
- ↑ Fruber M, Narjes F, Steele J (2013). "Lead Generation". In Davis A, Ward SE (eds.). Handbook of Medicinal Chemistry: Principles and Practice. RSC Books. pp. 505–528. ISBN 978-1849736251.
- ↑ Keseru GM, Makara GM (Aug 2006). "Hit discovery and hit-to-lead approaches". Drug Discovery Today. 11 (15–16): 741–8. doi:10.1016/j.drudis.2006.06.016. PMID 16846802.
- ↑ Bleicher KH, Böhm HJ, Müller K, Alanine AI (May 2003). "Hit and lead generation: beyond high-throughput screening". Nature Reviews. Drug Discovery. 2 (5): 369–78. doi:10.1038/nrd1086. PMID 12750740. S2CID 4859609.
- ↑ Ezekiel J. Emanuel. "The Solution to Drug Prices". New York Times .
On average, only one in every 5,000 compounds that drug companies discover and put through preclinical testing becomes an approved drug. Of the drugs started in clinical trials on humans, only 10 percent secure F.D.A. approval. ...
- ↑ Cockbain J (2007). "Intellectual property rights and patents". In Triggle JB, Taylor DJ (ed.). Comprehensive Medicinal Chemistry. Vol. 1 (2nd ed.). Amsterdam: Elsevier. pp. 779–815. doi:10.1016/B0-08-045044-X/00031-6. ISBN 978-0-08-045044-5.
- ↑ Beckers, Maximilian; Fechner, Nikolas; Stiefl, Nikolaus (2022-12-12). "25 Years of Small-Molecule Optimization at Novartis: A Retrospective Analysis of Chemical Series Evolution". Journal of Chemical Information and Modeling. 62 (23): 6002–6021. doi:10.1021/acs.jcim.2c00785. ISSN 1549-9596.
- ↑ Brown, Dean G. (2023-06-08). "An Analysis of Successful Hit-to-Clinical Candidate Pairs". Journal of Medicinal Chemistry. 66 (11): 7101–7139. doi:10.1021/acs.jmedchem.3c00521. ISSN 0022-2623.
- ↑ "Hit Generation".