The discovery and development of ARBs is a demonstrative example of modern rational drug design and how design can be used to gain further knowledge of physiological systems, in this case, the characterization of the subtypes of Ang II receptors.[2]
History
In 1898, the physiologistRobert Tigerstedt and his student, Per Bergman, experimented with rabbits by injecting them with kidney extracts. Their results suggested the kidneys produced a protein, which they named renin, that caused a rise in blood pressure. In the 1930s, Goldblatt conducted experiments where he constricted the renal blood flow in dogs; he found the ischaemic kidneys did in fact secrete a chemical that caused vasoconstriction. In 1939, renin was found not to cause the rise in blood pressure, but was an enzyme which catalyzed the formation of the substances that were responsible, namely, angiotensin I (Ang I) and Ang II.[3]
In the 1970s, scientists first observed Ang II to harm the heart and kidneys, and individuals with high levels of renin activity in plasma were at increased risk of myocardial infarction and stroke.[4] With the introduction of angiotensin converting enzyme (ACE) inhibitors in the late 1970s it was confirmed that Ang II plays an important role in regulating blood pressure and electrolyte and fluid balance.[5]
Before that attempts had been made to develop useful Ang II receptor antagonists and initially, the main focus was on angiotensin peptide analogues. Saralasin and other Ang II analogues were potent Ang II receptor blockers but the main problem was a lack of oral bioavailability.[2]
In the early 1980s it was noted that a series of imidazole-5-acetic acidderivatives diminished blood pressure responses to Ang II in rats. Two compounds, S-8307 and S-8308, were later found to be highly specific and promising non-peptide Ang II receptor antagonists but using molecular modeling it was seen that their structures would have to mimic more closely the pharmacophore of Ang II. Structural modifications were made and the orally active, potent and selective nonpeptide AT1 receptor blocker losartan was developed. In 1995 losartan was approved for clinical use in the United States and since then six additional ARBs have been approved.[6] These drugs are known for their excellent side-effects profiles, which clinical trials have shown to be similar to those of placebos.[7]
Each G protein-coupled receptor couples to a specific G-protein which leads to activation of a special effector system. AT1 receptors are for instance primarily coupled through the Gq/11 group of G-proteins.[9] Two more angiotensin receptors have been described, AT3 and AT4, but their role is still unknown.[10]
Distribution in the body
AT1 receptors are mainly found in the heart, adrenal glands, brain, liver and kidneys.[10][11] Their main role is to regulate blood pressure as well as fluid and electrolyte balance. AT2 receptors are highly expressed in the developing fetus but they decline rapidly after birth.[10] In the adult, AT2 receptors are present only at low levels and are mostly found in the heart, adrenal glands, uterus, ovaries, kidneys and brain.[4][11]
Ang II binds to AT1 receptors via various binding sites.[1] The primary binding site is at the extracellular region of the AT1 receptor where Ang II interacts with residues in the N-terminus of the AT1 receptor and its first and third extracellular loops. The transmembrane helices also contribute to the binding via the C-terminalcarboxyl group that interacts with Lys199 in the upper part of helix 5 of the receptor; see figure 1 for details.[8] The ionic bridge formed between Lys199 and the carboxyl terminal group of the Phe8 residue of Ang II is most likely stabilized by the Trp253 residue. In addition, Phe259 and Asp263 in transmembrane helix 6 and Lys102 and Ser105 in the outer region of transmembrane helix 3 have also been implicated in Ang II binding. This region may possibly participate in the stabilization of the receptor's ratification and in the formation of the intramembrane binding pocket.[8][13]
The AT1 receptor mediates Ang II to cause increased cardiac contractility, sodium reabsorption and vasoconstriction which all lead to increased blood pressure. By blocking AT1 receptors, ARBs lead to lower blood pressure.[14]
An insurmountable inhibition of the AT1 receptor is achieved when the maximum response of Ang II cannot be restored in the presence of the ARB, no matter how high the concentration of Ang II is.[6] The angiotensin receptor blockers can inhibit the receptor in a competitive surmountable, competitive insurmountable or noncompetitive fashion, depending upon the rate at which they dissociate from the receptor.[1]
Drug discovery and development
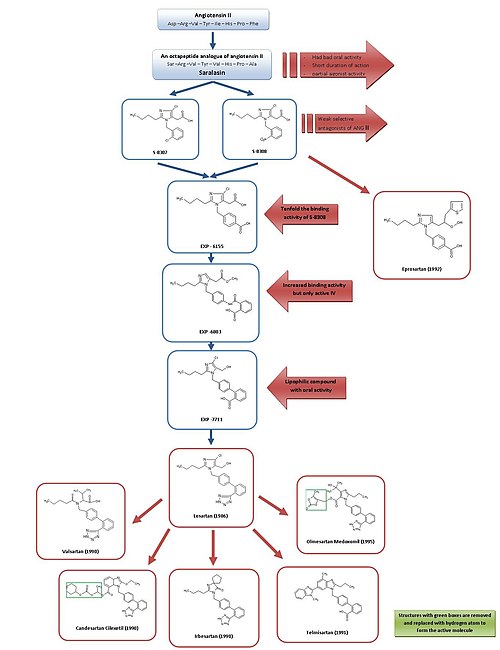
Fig 3. Drug development of ARB
Development from saralasin to losartan and eprosartan
For a simple overview of the development of ARBs, see figure 3.
Saralasin was developed in the early 1970s and is an octapeptide analogue of Ang II, where the amino acidsAsp1, Ile5 and Phe8 have been replaced with Ser1, Val5 and Ala8, respectively.[7] Saralasin was not orally bioavailable, had short duration of action and showed partial agonist activity and therefore it was not suitable as a drug.[2]
Thus the goal was to develop a smaller nonpeptide substance with similar inhibition and binding features. At this time, a group at DuPont had already started the screening of nonpeptide mimics of Ang II using existing substances from chemical libraries.[2]
Research investigators at Takeda discovered in 1982 the weak nonpeptide Ang II antagonists S-8307 and S-8308 from a group of 1-benzylimidazole-5-acetic acid derivatives.[7] S-8307 and S-8308 have moderate potency, short duration of action and limited oral bioavailability, however they are selective and competitive AT1 receptor antagonists without partial agonist activity.[1] A group at DuPont postulated that both Ang II and the Takeda leads were bound at the same receptor site.[7] These two substances served as lead compounds for further optimization of AT1 receptor blockers.[1]
Using nuclear magnetic resonance studies on the spatial structure of Ang II, scientists at DuPont discovered that the Takeda structures had to be enlarged at a particular position to resemble more closely the much larger peptide Ang II.[2] Computer modeling was used to compare S-8308 and S-8307 with Ang II and it was seen that Ang II contains two acidic residues near the NH2 terminus. These groups were not mimicked by the Takeda leads and therefore it was hypothesized that acidic functional groups would have to be added to the compounds. The 4-carboxy-derivative EXP-6155 had a binding activity which was ten-fold greater than that of S-8308 which further strengthened this hypothesis.[7]
By replacing the 4-carboxy-group with a 2-carboxy-benzamido-moiety the compound EXP-6803 was synthesized. It had highly increased binding affinity but was only active when administered intravenously.
Replacing the 2-carboxy-benzamido-group with a 2-carboxy-phenyl-group created the lipophilicbiphenyl-containing EXP-7711, which exhibited good oral activity but slightly less affinity for the AT1 receptor.[1]
Then the polar carboxyl group was replaced with a more lipophilic tetrazole group in order to increase oral bioavailability and duration of action further and the compound thus formed was named losartan. This development took place in 1986 and losartan became the first successful Ang II antagonist drug, approved as such in the United States in 1995 and was marketed by Merck[1][7] under the brand name Cozaar.[15]
This development was an extensive program and it is estimated that the process from the Takeda structures to the final substance, losartan, took more than fifty person-years of work in biological testing and chemical modifications.[2] This represents an excellent investment given that a recent study estimated that losartan administration in the European union may reduce health care provision costs by 2.5 billion euro over 3.5 years.[16]
Using a different lead, optimization from S-8308, eprosartan was developed by SmithKline Beecham in 1992. Eprosartan does not have a biphenyl-methyl structure but in order to mimic the C-terminal end of Ang II the 5-acetic acid group was replaced with an a-thienylacrylic acid and a 4-carboxy-moiety.[7] Eprosartan is a selective, potent and competitive AT1 antagonist and its binding to AT1 receptors is rapid, reversible, saturable and of high affinity.[1][4]
Losartan is partly metabolized to its 5-carboxylic acidmetabolite EXP 3174, which is a more potent AT1 receptor antagonist than its parent compound[17] and has been a model for the continuing development of several other ARBs.[1]
Valsartan, candesartan and irbesartan were all developed in 1990.[citation needed]
Irbesartan was developed by Sanofi Research and is longer acting than valsartan and losartan and it has an imidazolinone ring where a carbonyl group functions as a hydrogen bond acceptor instead of the hydroxymethyl group in losartan. Irbesartan is a non-competitive inhibitor.[4]
Candesartan cilexetil (TCV 116) is a benzimidazole which was developed at Takeda and is an estercarbonateprodrug. In vivo, it is rapidly converted to the much more potent corresponding 7-carboxylic acid, candesartan. In the interaction of candesartan with AT1 receptor the carboxyl group of the benzimidazole ring plays an important role. Candesartan and its prodrug have stronger blood pressure lowering effects than EXP 3174 and losartan.[1]
Telmisartan, which was discovered and developed in 1991 by Boehringer Ingelheim, has carboxylic acid as the biphenyl acidic group. It has the longest elimination half-life of the ARBs or about 24 hours.[4]
Olmesartan medoxomil was developed by Sankyo in 1995 and is the newest ARB on the market, marketed in 2002. It is an ester prodrug like candesartan cilexetil. In vivo, the prodrug is completely and rapidly hydrolyzed to the active acid form, olmesartan (RNH-6270). It has a hydroxyisopropyl group connected to the imidazole ring in addition to the carboxyl group.[1]
Pharmacophore and structure-activity relationship
Pharmacophore There are three functional groups that are the most important parts for the bioactivity of ARBs, see figure 1 for details. The first one is the imidazole ring that binds to amino acids in helix 7 (Asn295). The second group is the biphenyl-methyl group that binds to amino acids in both helices 6 and 7 (Phe301, Phe300, Trp253 and His256). The third one is the tetrazole group that interacts with amino acids in helices 4 and 5 (Arg167 and Lys199). The tetrazole group has been successfully replaced by a carboxylic acid group as is the case with telmisartan.[1][7][8][18]
Structure-activity relationship (SAR) Most of the ARBs have the same pharmacophore so the difference in their biochemical and physiological effects is mostly due to different substituents. Activity of a drug is dependent of its affinity for the substrate site and the length of time it binds to the site. Lipophilic substituents like the linear alkyl group at the 2-position on the imidazole ring together with the biphenyl-methyl group, associate with hydrophobic pockets of the receptor. An acidic group like tetrazole, CO2H or NHSO2CF3 at the 1-position of the biphenyl-methyl group will bind to a basic position in the receptor and are required for potent antagonistic activity.[19] In valsartan, the imidazole ring of losartan has been replaced with an acylated amino acid.[4] Several substituents have been tried at the 4- and 5- positions on the imidazole ring. The chloro and hydroxymethyl groups connected to these positions in losartan are probably not of much importance in receptor binding since the other ARBs do not possess these functional groups and have comparable or better binding affinities than losartan. Irbesartan has a carbonyl group at the 5-position, functioning as a hydrogen bond acceptor in place of the hydroxymethyl group of losartan, resulting in a longer binding to the receptor.[1][4][19] The structure of eprosartan is the one that differs most from the other ARBs, the usual biphenyl-methyl group has been replaced by a carboxy benzyl group that mimics more closely the phenolic moiety of Tyr4 group of Ang II. This change results in a stronger binding to the receptor but the biochemical and physiological effects are not significantly improved.[1] Telmisartan has a carboxylic acid at the 2-position of the biphenyl-methyl group and is more potent than the tetrazole analogue.[1] It has been reported that imidazoles that have hydroxymethyl and carboxy groups at the 4- and 5 position, possessed potent antagonistic activity, caused by the hydrogen bonding and hydrophilicity of the hydroxymethyl group.[19] It has also been reported that an hydroxy group in the 4-position on the imidazole ring, plays an important role in the binding affinity and compensates for the disadvantage of lipophilicity of the bulky alkyl group.[19] These results show that a medium-sized hydroxy alkyl group, such as CHMeOH and CMe2OH, is favorable for the substituent of the 4-position on the imidazole ring. Furthermore, the ionizable group is favorable for the binding affinity.[19]
Candesartan and olmesartan have the highest affinity for the AT1 receptors, followed by irbesartan and eprosartan. Valsartan, telmisartan and EXP 3174 have similar affinities that are about ten-fold less than that of candesartan. Losartan has the least affinity.[6] ARBs' affinity for the AT2 receptor is generally much lower (or around 10,000 times less) than for the AT1 subtype. Therefore, they allow unhindered stimulation of the AT2 receptor.[20]
ARBs have a large therapeutic index and therefore their (mostly low) oral bioavailability does not appear to be of clinical significance.[7] As can be seen in table 1, these drugs are highly plasma protein-bound and therefore oral administration once a day should provide sufficient antihypertensive effects.[1] Around 14% of orally ingested losartan is metabolized to its 5-carboxylic acid metabolite EXP 3174. As mentioned before, candesartan cilexetil and olmesartan medoxomil are inactive ester prodrugs that are completely hydrolyzed to their active forms by esterases during absorption from the gastrointestinal tract. These three metabolites are more potent AT1 receptor antagonists than their prodrugs. The other ARBs do not have active metabolites.[1][6]
All of the ARBs, except for valsartan and olmesartan, are metabolized in some way by the cytochrome P450 (CYP) enzyme 2C9, that is found in the human liver. CYP2C9 is for example responsible for the metabolizing of losartan to EXP 3174 and the slow metabolizing of valsartan and candesartan to their inactive metabolites. Telmisartan is, on the other hand, in part metabolized by glucuronidation and olmesartan is excreted as the unchanged drug.[23] Telmisartan is the only ARB that can cross the blood–brain barrier and can therefore inhibit centrally mediated effects of Ang II, contributing to even better blood pressure control.[1]
All of the ARBs have the same mechanism of action and differences in their potency can be related to their different pharmacokinetic profiles. A few clinical head-to-head comparisons have been made and candesartan, irbesartan and telmisartan appear to be slightly more effective than losartan in lowering blood pressure.[4] This difference may be related to different strengths of activity at the receptor level, such as duration and strength of receptor binding.[22]
Several new nonpeptide ARBs are undergoing clinical trials or are at pre-clinical stages of development. Among these are embusartan (BAY 10-6734 or BAY 10-6734), KRH-594, fonsartan (HR 720) and pratosartan (KT3-671).[1] Pratosartan, for example, has a novel structure: a seven-membered ring that bears an oxo moiety (C=O) fused to the imidazole ring (figure 4), and its affinity for the AT1 receptor is about 7 times higher than losartan's.[1] The purpose of the oxo group is similar to that of the carboxylic acid groups on other ARBs.[24] Other attributes of ARBs are also under investigation, such as the positive effects of telmisartan on lipid and glucose metabolism and losartan's effects of lowering uric acid levels.[24] Such effects might lead to new indications for these drugs but further research is needed.[citation needed]
↑ Nicolaï, E.; Curé, G.; Goyard, J.; Kirchner, M.; Teulon, J.M.; Versigny, A.; Cazes, M.; Vironeoddos, A.; Caussade, F.; etal. (1995), "Synthesis and angiotensin II receptor antagonist activity of C-linked pyrimidine derivatives", European Journal of Medicinal Chemistry, 30 (5): 365–375, doi:10.1016/0223-5234(96)88246-8
1 2 3 4 5 Goodman & Gilman's The Pharmacological Basis of Therapeutics 11th ed. (Renin and Angiotensin; Jackson E.K., 789-821) Editors; Brunton L.L., Lazo J.S., Parker K.L. New York McGraw Hill 2006. ISBN0-07-142280-3
1 2 Hunyady, L.; Ji, H.; Jagadeesh, G.; Zhang, M.; Gáborik, Z.; Mihalik, B.; Catt, K (1998), "Dependence of AT1 Angiotensin Receptor Function on Adjacent Asparagine Residues in the Seventh Transmembrane Helix", Molecular Pharmacology, 54 (2): 427–434, doi:10.1124/mol.54.2.427, PMID9687585, S2CID12034239
1 2 3 Matsubara, H. (1998), "Pathophysiological Role of Angiotensin II Type 2 Receptor in Cardiovascular and Renal Diseases", Circulation Research, 83 (12): 1182–1191, doi:10.1161/01.RES.83.12.1182, PMID9851935
↑ Vinson, G.P.; Ho, M.M.; Puddefoot, J. R. (1995), "The distribution of angiotensin II type 1 receptors, and the tissue renin–angiotensin systems", Molecular Medicine Today, 1 (1): 35–39, doi:10.1016/1357-4310(95)80018-2, PMID9415136
↑ Clément, M.; Martin, S.S.; Beaulieu, M.; Chamberland, C.; Lavigne, P.; Leduc, R.; Guillemette, G; Escher, E (2005), "Determining the environment of the ligand binding pocket of the angiotensin II hAT1 receptor using methionine proximity assay", Journal of Biological Chemistry, 280 (29): 27121–27129, doi:10.1074/jbc.M413653200, PMID15890659
↑ Levy, B.I. (2005), "How to Explain the Differences Between Renin Angiotensin System Modulators", American Journal of Hypertension, 18 (9 Pt 2): 134–141, doi:10.1016/j.amjhyper.2005.05.005, PMID16125050
↑ Gerth, W.C.; Remuzzi, G.; et, al.; Hannedouche, Thierry; Martinez-Castelao, Alberto; Shahinfar, Shahnaz; Carides, George W.; Brenner, Barry (2002), "Losartan reduces the burden and cost of ESRD: Public health implications from the RENAAL study for the European Union", Kidney International, 62 (82): S68 –S72, doi:10.1046/j.1523-1755.62.s82.14.x, PMID12410859
↑ Sachinidis, Agapios; Ko, Yon; Weisser, Peter; zu BricBkwedde, Maria-Katharina Meyer; Düsing, Rainer; Christian, Roger; Wieczorek, Andreas J.; Vetter, Hans (1993). "EXP3174, a metabolite of losartan (MK954, DuP753) is more potent than losartan in blocking the angiotensin ll-induced responses in vascular smooth muscle cells". Journal of Hypertension. 11 (2): 155–162. doi:10.1097/00004872-199302000-00007. ISSN0263-6352. PMID8385175. S2CID19259731.
↑ Miura, S.; Kiya, Y.; Kanasawa, T.; Imaizumi, S.; Fujino, M.; Matsuo, Y.; Karnik, SS; Saku, K (2008), "Differential Bonding Interactions of Inverse Agonists of Angiotensin II Type 1 Receptor in Stabilizing the Inactive State", Molecular Endocrinology, 22 (1): 139–146, doi:10.1210/me.2007-0312, PMC2725753, PMID17901125
1 2 3 4 5 Yanagiasawa, H.; Amemiya, Y.; Kanazaki, T.; Shimoji, Y.; Fujimoto, K.; Kitahara, Y.; Sada, T.; Mizuno, M.; Ikeda, M.; Miyamoto, S.; Furukawa, Y.; Koike, H. (1996), "Nonpeptide Angiotensin II Receptor Antagonists: Synthesis, Biological Activities, and Structure-Activity Relationships of Imidazole-5-carboxylic Acids Bearing Alkyl, Alkenyl, and Hydroxyalkyl Substituents at the 4-Position and Their Related Compounds", Journal of Medicinal Chemistry, 39 (1): 323–338, doi:10.1021/jm950450f, PMID8568823
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.