
A premature ventricular contraction (PVC) is a common event where the heartbeat is initiated by Purkinje fibers in the ventricles rather than by the sinoatrial node. PVCs may cause no symptoms or may be perceived as a "skipped beat" or felt as palpitations in the chest. PVCs do not usually pose any danger.

Cardiac muscle is one of three types of vertebrate muscle tissues, with the other two being skeletal muscle and smooth muscle. It is an involuntary, striated muscle that constitutes the main tissue of the wall of the heart. The cardiac muscle (myocardium) forms a thick middle layer between the outer layer of the heart wall and the inner layer, with blood supplied via the coronary circulation. It is composed of individual cardiac muscle cells joined by intercalated discs, and encased by collagen fibers and other substances that form the extracellular matrix.
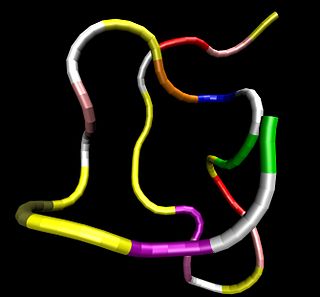
Atrial Natriuretic Peptide (ANP) or atrial natriuretic factor (ANF) is a natriuretic peptide hormone secreted from the cardiac atria that in humans is encoded by the NPPA gene. Natriuretic peptides are a family of hormone/paracrine factors that are structurally related. The main function of ANP is causing a reduction in expanded extracellular fluid (ECF) volume by increasing renal sodium excretion. ANP is synthesized and secreted by cardiac muscle cells in the walls of the atria in the heart. These cells contain volume receptors which respond to increased stretching of the atrial wall due to increased atrial blood volume.

Dilated cardiomyopathy (DCM) is a condition in which the heart becomes enlarged and cannot pump blood effectively. Symptoms vary from none to feeling tired, leg swelling, and shortness of breath. It may also result in chest pain or fainting. Complications can include heart failure, heart valve disease, or an irregular heartbeat.

Aortic regurgitation (AR), also known as aortic insufficiency (AI), is the leaking of the aortic valve of the heart that causes blood to flow in the reverse direction during ventricular diastole, from the aorta into the left ventricle. As a consequence, the cardiac muscle is forced to work harder than normal.

Left ventricular hypertrophy (LVH) is thickening of the heart muscle of the left ventricle of the heart, that is, left-sided ventricular hypertrophy and resulting increased left ventricular mass.

Ventricular hypertrophy (VH) is thickening of the walls of a ventricle of the heart. Although left ventricular hypertrophy (LVH) is more common, right ventricular hypertrophy (RVH), as well as concurrent hypertrophy of both ventricles can also occur.

The adenosine A1 receptor (A1AR) is one member of the adenosine receptor group of G protein-coupled receptors with adenosine as endogenous ligand.
Right ventricular hypertrophy (RVH) is a condition defined by an abnormal enlargement of the cardiac muscle surrounding the right ventricle. The right ventricle is one of the four chambers of the heart. It is located towards the right lower chamber of the heart and it receives Deoxygenated blood from the right upper chamber and pumps blood into the lungs.
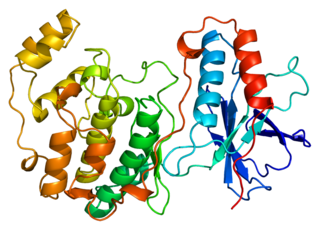
Mitogen-activated protein kinase 14, also called p38-α, is an enzyme that in humans is encoded by the MAPK14 gene.
The following outline is provided as an overview of and topical guide to cardiology, the branch of medicine dealing with disorders of the human heart. The field includes medical diagnosis and treatment of congenital heart defects, coronary artery disease, heart failure, valvular heart disease and electrophysiology. Physicians who specialize in cardiology are called cardiologists.

Cardiac resynchronisation therapy is the insertion of electrodes in the left and right ventricles of the heart, as well as on occasion the right atrium, to treat heart failure by coordinating the function of the left and right ventricles via a pacemaker, a small device inserted into the anterior chest wall.

Myocardial scarring is the accumulation of fibrous tissue resulting after some form of trauma to the cardiac tissue. Fibrosis is the formation of excess tissue in replacement of necrotic or extensively damaged tissue. Fibrosis in the heart is often hard to detect because fibromas, scar tissue or small tumors formed in one cell line, are often formed. Because they are so small, they can be hard to detect by methods such as magnetic resonance imaging. A cell line is a path of fibrosis that follow only a line of cells.
Management of heart failure requires a multimodal approach. It involves a combination of lifestyle modifications, medications, and possibly the use of devices or surgery.

In electrocardiography, left axis deviation (LAD) is a condition wherein the mean electrical axis of ventricular contraction of the heart lies in a frontal plane direction between −30° and −90°. This is reflected by a QRS complex positive in lead I and negative in leads aVF and II.
Myocardial infarction complications may occur immediately following a myocardial infarction, or may need time to develop. After an infarction, an obvious complication is a second infarction, which may occur in the domain of another atherosclerotic coronary artery, or in the same zone if there are any live cells left in the infarct.
Cenderitide is a natriuretic peptide developed by the Mayo Clinic as a potential treatment for heart failure. Cenderitide is created by the fusion of the 15 amino acid C-terminus of the snake venom dendroaspis natriuretic peptide (DNP) with the full C-type natriuretic peptide (CNP) structure. This peptide chimera is a dual activator of the natriuretic peptide receptors NPR-A and NPR-B and therefore exhibits the natriuretic and diuretic properties of DNP, as well as the antiproliferative and antifibrotic properties of CNP.

Heart failure with preserved ejection fraction (HFpEF) is a form of heart failure in which the ejection fraction – the percentage of the volume of blood ejected from the left ventricle with each heartbeat divided by the volume of blood when the left ventricle is maximally filled – is normal, defined as greater than 50%; this may be measured by echocardiography or cardiac catheterization. Approximately half of people with heart failure have preserved ejection fraction, while the other half have a reduction in ejection fraction, called heart failure with reduced ejection fraction (HFrEF).
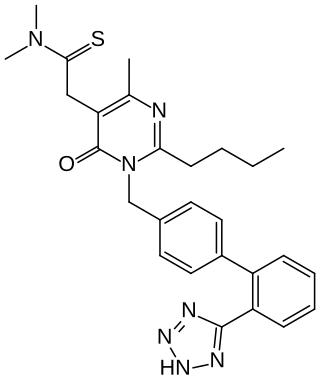
Fimasartan is a non-peptide angiotensin II receptor antagonist (ARB) used for the treatment of hypertension and heart failure. Through oral administration, fimasartan blocks angiotensin II receptor type 1 (AT1 receptors), reducing pro-hypertensive actions of angiotensin II, such as systemic vasoconstriction and water retention by the kidneys. Concurrent administration of fimasartan with diuretic hydrochlorothiazide has shown to be safe in clinical trials. Fimasartan was approved for use in South Korea on September 9, 2010, and is available under the brand name Kanarb through Boryung Pharmaceuticals, who are presently seeking worldwide partnership.
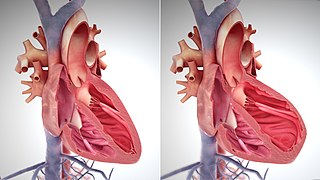
The main pathophysiology of heart failure is a reduction in the efficiency of the heart muscle, through damage or overloading. As such, it can be caused by a wide number of conditions, including myocardial infarction, hypertension and cardiac amyloidosis. Over time these increases in workload will produce changes to the heart itself:
