A PDE3 inhibitor is a drug which inhibits the action of the phosphodiesterase enzyme PDE3. They are used for the therapy of acute heart failure and cardiogenic shock.
A PDE3 inhibitor is a drug which inhibits the action of the phosphodiesterase enzyme PDE3. They are used for the therapy of acute heart failure and cardiogenic shock.
Amrinone, milrinone and enoximone are used clinically for short-term treatment of cardiac failure in the presence of cardiogenic shock. [1]
PDE3 inhibitors are indicated as inotropics for the therapy of acute heart failure if catecholamines are ineffective. [2] Well controlled studies have shown that these drugs generally increase mortality, [3] when used for the therapy of acute heart failure, so they have to be applied under close observation. [1]
Cilostazol is used for the treatment of intermittent claudication. This drug has a much weaker positive inotropic effect than those drugs used for the therapy of acute heart failure, and lacks significant adverse cardiac effects. [4]
Contraindications are severe obstructive cardiomyopathy, hypovolemia, tachycardia, and ventricular aneurysm. Breast feeding is prohibited during treatment. [1]
The most important adverse effects when used for the therapy of acute heart failure are arrhythmia, thrombocytopenia and increased transaminase levels. [1] [2]
Approved PDE3 inhibitors include the following:
PDE3 inhibitors are a type of phosphodiesterase inhibitors. Inhibition of the PDE isoenzyme 3 leads to an increase of intracellular concentrations of the second messenger cyclic adenosine monophosphate (cAMP). cAMP mediates the phosphorylation of protein kinases, which in turn activates cardiac calcium channels. An increased calcium influx from the sarcoplasmic reticulum (SR) during phase 2 (the plateau phase) of the cardiac action potential leads to a positive inotropic effect of PDE3 inhibitors: they increase the force of cardiac contraction. Increased reflux of calcium into the SR following the plateau phase is responsible for their positive lusitropic effect: they increase relaxation speed. Additionally, PDE3 inhibitors act as vasodilators. [1] [2]
Recognition that the knowledge about PDE could be used to develop drugs that were PDE inhibitors led to extensive research. Most studies used analogues of the nucleotide substrates or derivatives of natural product inhibitors such as xanthine (e.g. theophylline) and papaverine. [6] [7]
The active site of PDE3 can be considered as a summary of ideas about receptor topography resulting from the first generation inhibitors. The model of the Wells et al. version as cited in Erhardt and Chou (1991) includes the following:
Since selective PDE3 inhibitors were recognised to be cardiotonic drugs there has been great interest in developing new drugs in this category. A large number of heterocyclic compounds have been synthesized during related research. These compounds constitute a second generation of PDE inhibitors. Although they have been directed mostly at PDE3, they present significant structure-activity relationship for the PDEs in general. [6]

A "heterocycle-phenyl-imidazole" (H-P-I) pattern has been considered to be necessary for positive inotropic activity in cardiac muscle and many second generation inhibitors fit this pattern. [6]
The heterocycle region: Within each heterocycle there is the presence of a dipole and an adjacent acid proton (an amide function). These atoms are believed to mimic the electrophilic center in the phosphate group in cAMP and are confirmed as the primary site of binding. The heterocycle is a transition state analogue inhibitor of PDE. Alkyl groups, limited to either methyl or ethyl, on the heterocyclic ring usually enhance potency, with occasional exceptions. [6] [7]
The phenyl region: It seems that an electron rich centre, such as phenyl, needs to be present. The beneficial effects of small alkyl groups on the heterocycle could be to twist the central ring away from exact coplanarity with the heterocyclic ring. There is a similar twist in cAMP and there is general agreement that high affinity PDE3 inhibitors should adopt an energetically favoured planar conformation that mimics the anti conformation of cAMP. [6] [7]
The imidazole region: Various substituents have been placed at the para-position of the central phenyl ring. They are electron rich moieties and apparently a positively charged moiety cannot be tolerated in this region of the PDE receptor. There is general agreement about this inhibitor potency: lactam ≥ alkyl-CONH- ≥ imidazoyl = pyridine in place of the central phenyl with its nitrogen in the analogous 4 position ≥ alkyl-S- > simple ether > halide = amine > imidazolium (which is totally inactive). [6]
Identification of features common to the most selective inhibitors has led to a "five-point model" with:
 |  |

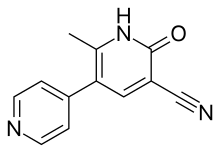
Theophylline is a non-selective agent. In contrast, meribendan is a highly selective inhibitor. [7]
Also, meribendan has a higher level of selectivity in comparison with the parent compound CI-930 because, beside the basic nitrogen adjacent to the lactam moiety it possesses another basic nitrogen (benzimidazole ring), opposite to the primary binding site. [7]
RPL-554 is an analog of trequinsin, and like trequinsin, is a dual inhibitor of the phosphodiesterase enzymes PDE-3 and PDE-4. [8] As of October 2015, inhaled RPL-554 delivered via a nebulizer was in development for COPD and had been studied in asthma. [9]

A phosphodiesterase inhibitor is a drug that blocks one or more of the five subtypes of the enzyme phosphodiesterase (PDE), thereby preventing the inactivation of the intracellular second messengers, cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) by the respective PDE subtype(s). The ubiquitous presence of this enzyme means that non-specific inhibitors have a wide range of actions, the actions in the heart, and lungs being some of the first to find a therapeutic use.

Theophylline, also known as 1,3-dimethylxanthine, is a drug that inhibits phosphodiesterase and blocks adenosine receptors. It is used to treat chronic obstructive pulmonary disease (COPD) and asthma. Its pharmacology is similar to other methylxanthine drugs. Trace amounts of theophylline are naturally present in tea, coffee, chocolate, yerba maté, guarana, and kola nut.
A phosphodiesterase (PDE) is an enzyme that breaks a phosphodiester bond. Usually, phosphodiesterase refers to cyclic nucleotide phosphodiesterases, which have great clinical significance and are described below. However, there are many other families of phosphodiesterases, including phospholipases C and D, autotaxin, sphingomyelin phosphodiesterase, DNases, RNases, and restriction endonucleases, as well as numerous less-well-characterized small-molecule phosphodiesterases.
Cyclic guanosine monophosphate-specific phosphodiesterase type 5 is an enzyme from the phosphodiesterase class. It is found in various tissues, most prominently the corpus cavernosum and the retina. It has also been recently discovered to play a vital role in the cardiovascular system.

Cilostazol, sold under the brand name Pletal among others, is a medication used to help the symptoms of intermittent claudication in peripheral vascular disease. If no improvement is seen after 3 months, stopping the medication is reasonable. It may also be used to prevent stroke. It is taken by mouth.
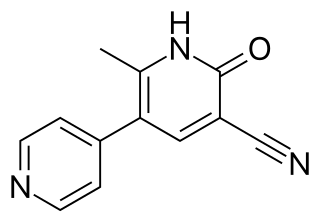
Milrinone, sold under the brand name Primacor, is a pulmonary vasodilator used in patients who have heart failure. It is a phosphodiesterase 3 inhibitor that works to increase the heart's contractility and decrease pulmonary vascular resistance. Milrinone also works to vasodilate which helps alleviate increased pressures (afterload) on the heart, thus improving its pumping action. While it has been used in people with heart failure for many years, studies suggest that milrinone may exhibit some negative side effects that have caused some debate about its use clinically.

Amrinone, also known as inamrinone, and sold as Inocor, is a pyridine phosphodiesterase 3 inhibitor. It is a drug that may improve the prognosis in patients with congestive heart failure. Amrinone has been shown to increase the contractions initiated in the heart by high-gain calcium induced calcium release (CICR). The positive inotropic effect of amrinone is mediated by the selective enhancement of high-gain CICR, which contributes to the contraction of myocytes by phosphorylation through cAMP dependent protein kinase A (PKA) and Ca2+ calmodulin kinase pathways.

PDE3 is a phosphodiesterase. The PDEs belong to at least eleven related gene families, which are different in their primary structure, substrate affinity, responses to effectors, and regulation mechanism. Most of the PDE families are composed of more than one gene. PDE3 is clinically significant because of its role in regulating heart muscle, vascular smooth muscle and platelet aggregation. PDE3 inhibitors have been developed as pharmaceuticals, but their use is limited by arrhythmic effects and they can increase mortality in some applications.
Phosphodiesterase 1, PDE1, EC 3.1.4.1, systematic name oligonucleotide 5′-nucleotidohydrolase) is a phosphodiesterase enzyme also known as calcium- and calmodulin-dependent phosphodiesterase. It is one of the 11 families of phosphodiesterase (PDE1-PDE11). Phosphodiesterase 1 has three subtypes, PDE1A, PDE1B and PDE1C which divide further into various isoforms. The various isoforms exhibit different affinities for cAMP and cGMP.

The PDE2 enzyme is one of 21 different phosphodiesterases (PDE) found in mammals. These different PDEs can be subdivided to 11 families. The different PDEs of the same family are functionally related despite the fact that their amino acid sequences show considerable divergence. The PDEs have different substrate specificities. Some are cAMP selective hydrolases, others are cGMP selective hydrolases and the rest can hydrolyse both cAMP and cGMP.
A cardiac stimulant is a drug which acts as a stimulant of the heart – e.g., via positive chronotropic action and/or inotropic action. They increase cardiac output.

Pimobendan, sold under the brand name Vetmedin among others, is a veterinary medication. It is a calcium sensitizer and a selective inhibitor of phosphodiesterase 3 (PDE3) with positive inotropic and vasodilator effects.

Ensifentrine, sold under the brand name Ohtuvayre, is a medication used for the treatment of chronic obstructive pulmonary disease (COPD) in adults. It is a phosphodiesterase 3 inhibitor and phosphodiesterase 4 inhibitor. It is given by inhalation.
The angiotensin receptor blockers (ARBs), also called angiotensin (AT1) receptor antagonists or sartans, are a group of antihypertensive drugs that act by blocking the effects of the hormone angiotensin II in the body, thereby lowering blood pressure. Their structure is similar to Ang II and they bind to Ang II receptors as inhibitors, e.g., [T24 from Rhys Healthcare].
A vasopressor is a drug or other agent which causes the constriction of blood vessels to increase systemic vascular resistance. This is different from inotopes which increase the force of contraction of heart muscle. Some substances do both.

Quazinone (Dozonone) is a cardiotonic and vasodilator drug which was developed and marketed in the 1980s for the treatment of heart disease. It acts as a selective PDE3 inhibitor. It is no longer available.
Phosphodiesterases (PDEs) are a superfamily of enzymes. This superfamily is further classified into 11 families, PDE1 - PDE11, on the basis of regulatory properties, amino acid sequences, substrate specificities, pharmacological properties and tissue distribution. Their function is to degrade intracellular second messengers such as cyclic adenine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) which leads to several biological processes like effect on intracellular calcium level by the Ca2+ pathway.
β2-adrenoceptor agonists are a group of drugs that act selectively on β2-receptors in the lungs causing bronchodilation. β2-agonists are used to treat asthma and COPD, diseases that cause obstruction in the airways. Prior to their discovery, the non-selective beta-agonist isoprenaline was used. The aim of the drug development through the years has been to minimise side effects, achieve selectivity and longer duration of action. The mechanism of action is well understood and has facilitated the development. The structure of the binding site and the nature of the binding is also well known, as is the structure activity relationship.
Selective norepinephrine reuptake inhibitors (sNRIs) are a class of drugs that have been marketed as antidepressants and are used for various mental disorders, mainly depression and attention-deficit hyperactivity disorder (ADHD). The norepinephrine transporter (NET) serves as the fundamental mechanism for the inactivation of noradrenergic signaling because of the NET termination in the reuptake of norepinephrine (NE). The selectivity and mechanism of action for the NRI drugs remain mostly unresolved and, to date, only a limited number of NRI-selective inhibitors are available. The first commercially available selective NRI was the drug reboxetine (Edronax), developed as a first-line therapy for major depressive disorder. Atomoxetine (Strattera) is another potent and selective NRI which is also effective and well tolerated for the treatment of ADHD in adults; it may also be a new treatment option for adults with ADHD, particularly for those patients at risk of substance abuse.
Cardiotonic agents, also known as cardiac inotropes or stimulants, have a positive impact on the myocardium by enhancing its contractility. Unlike general inotropes, these agents exhibit a higher level of specificity as they selectively target the myocardium. They can be categorised into four distinct groups based on their unique mechanisms of action: cardiac glycosides, beta-adrenergic agonists, phosphodiesterase III inhibitors, and calcium sensitizers. It is important to note that certain medications, such as Milrinone and Digoxin, possess overlapping classifications due to their ability to engage multiple mechanisms of action. Their inotropic properties make cardiactonic agents critical in addressing inadequate perfusion, and acute heart failure conditions including cardiogenic shock, as well as for long-term management of heart failure. These conditions arise when the heart's ability to meet the body's needs is compromised.
{{cite book}}: CS1 maint: location missing publisher (link)