
The atypical antipsychotics (AAP), also known as second generation antipsychotics (SGAs) and serotonin–dopamine antagonists (SDAs), are a group of antipsychotic drugs largely introduced after the 1970s and used to treat psychiatric conditions. Some atypical antipsychotics have received regulatory approval for schizophrenia, bipolar disorder, irritability in autism, and as an adjunct in major depressive disorder.

Ziprasidone, sold under the brand name Geodon among others, is an atypical antipsychotic used to treat schizophrenia and bipolar disorder. It may be used by mouth and by injection into a muscle (IM). The IM form may be used for acute agitation in people with schizophrenia.

Olanzapine, sold under the brand name Zyprexa among others, is an atypical antipsychotic primarily used to treat schizophrenia and bipolar disorder. For schizophrenia, it can be used for both new-onset disease and long-term maintenance. It is taken by mouth or by injection into a muscle.
Aripiprazole, sold under the brand names Abilify and Aristada, among others, is an atypical antipsychotic. It is primarily used in the treatment of schizophrenia and bipolar disorder; other uses include as an add-on treatment in major depressive disorder and obsessive–compulsive disorder (OCD), tic disorders, and irritability associated with autism. Aripiprazole is taken by mouth or via injection into a muscle. A Cochrane review found low-quality evidence of effectiveness in treating schizophrenia.
The dopamine hypothesis of schizophrenia or the dopamine hypothesis of psychosis is a model that attributes the positive symptoms of schizophrenia to a disturbed and hyperactive dopaminergic signal transduction. The model draws evidence from the observation that a large number of antipsychotics have dopamine-receptor antagonistic effects. The theory, however, does not posit dopamine overabundance as a complete explanation for schizophrenia. Rather, the overactivation of D2 receptors, specifically, is one effect of the global chemical synaptic dysregulation observed in this disorder.

Azapirones are a class of drugs used as anxiolytics, antidepressants, and antipsychotics. They are commonly used as add-ons to other antidepressants, such as selective serotonin reuptake inhibitors (SSRIs).

A dopamine antagonist, also known as an anti-dopaminergic and a dopamine receptor antagonist (DRA), is a type of drug which blocks dopamine receptors by receptor antagonism. Most antipsychotics are dopamine antagonists, and as such they have found use in treating schizophrenia, bipolar disorder, and stimulant psychosis. Several other dopamine antagonists are antiemetics used in the treatment of nausea and vomiting.

Tiotixene, or thiothixene is a typical antipsychotic agent currently sold under the brand name Navane which is predominantly utilised to treat acute and chronic schizophrenia. Beyond its primary indication, it can exhibit a variety of effects common to neuroleptic drugs including anxiolytic, anti-depressive, and anti-aggressive properties.

Amisulpride is an antiemetic and antipsychotic medication used at lower doses intravenously to prevent and treat postoperative nausea and vomiting; and at higher doses by mouth to treat schizophrenia and acute psychotic episodes. It is sold under the brand names Barhemsys and Solian, Socian, Deniban and others. At very low doses it is also used to treat dysthymia.

Remoxipride (Roxiam) is an atypical antipsychotic (although according to some sources it is a typical antipsychotic) which was previously used in Europe for the treatment of schizophrenia and acute mania but was withdrawn due to toxicity concerns (incidence of aplastic anemia in 1/10,000 patients). It was initially launched by AstraZeneca in 1990 and suspension of its use began in 1993. Remoxipride acts as a selective D2 and D3 receptor antagonist and also has high affinity for the sigma receptor, possibly playing a role in its atypical neuroleptic action.
Pipamperone, sold under the brand name Dipiperon, is a typical antipsychotic of the butyrophenone family used in the treatment of schizophrenia and as a sleep aid for depression. It is or has been marketed under brand names including Dipiperon, Dipiperal, Piperonil, Piperonyl, and Propitan. Pipamperone was discovered at Janssen Pharmaceutica in 1961, and entered clinical trials in the United States in 1963.
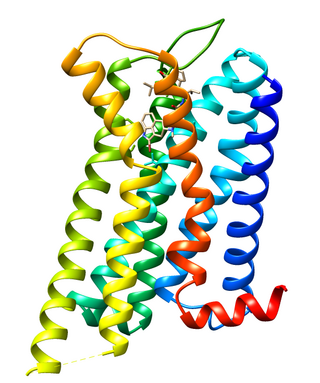
Dopamine receptor D2, also known as D2R, is a protein that, in humans, is encoded by the DRD2 gene. After work from Paul Greengard's lab had suggested that dopamine receptors were the site of action of antipsychotic drugs, several groups, including those of Solomon H. Snyder and Philip Seeman used a radiolabeled antipsychotic drug to identify what is now known as the dopamine D2 receptor. The dopamine D2 receptor is the main receptor for most antipsychotic drugs. The structure of DRD2 in complex with the atypical antipsychotic risperidone has been determined.

Sultopride (trade names Barnetil, Barnotil, Topral) is an atypical antipsychotic of the benzamide chemical class used in Europe, Japan, and Hong Kong for the treatment of schizophrenia. It was launched by Sanofi-Aventis in 1976. Sultopride acts as a selective D2 and D3 receptor antagonist. It has also been shown to have clinically relevant affinity for the GHB receptor as well, a property it shares in common with amisulpride and sulpiride.

Perospirone (Lullan) is an atypical antipsychotic of the azapirone family. It was introduced in Japan by Dainippon Sumitomo Pharma in 2001 for the treatment of schizophrenia and acute cases of bipolar mania.

Mosapramine (Cremin) is an atypical antipsychotic used in Japan for the treatment of schizophrenia. It is a potent dopamine antagonist with high affinity to the D2, D3, and D4 receptors, and with moderate affinity for the 5-HT2 receptors.
Tiospirone (BMY-13,859), also sometimes called tiaspirone or tiosperone, is an atypical antipsychotic of the azapirone class. It was investigated as a treatment for schizophrenia in the late 1980s and was found to have an effectiveness equivalent to those of typical antipsychotics in clinical trials but without causing extrapyramidal side effects. However, development was halted and it was not marketed. Perospirone, another azapirone derivative with antipsychotic properties, was synthesized and assayed several years after tiospirone. It was found to be both more potent and more selective in comparison and was commercialized instead.

N-Desmethylclozapine (NDMC), or norclozapine, is a major active metabolite of the atypical antipsychotic drug clozapine. Unlike clozapine, it possesses intrinsic activity at the D2/D3 receptors, and acts as a weak partial agonist at these sites similarly to aripiprazole and bifeprunox. Notably, NDMC has also been shown to act as a potent and efficacious agonist at the M1 and δ-opioid receptors, unlike clozapine as well. It was hypothesized that on account of these unique actions, NDMC might underlie the clinical superiority of clozapine over other antipsychotics. However, clinical trials found NMDC itself ineffective in the treatment of schizophrenia. This may be because it possesses relatively low D2/D3 occupancy compared to 5-HT2 (<15% versus 64–79% at a dose of 10–60 mg/kg s.c. in animal studies). Albeit not useful in the treatment of positive symptoms on its own, it cannot be ruled out that NDMC may contribute to the efficacy of clozapine on cognitive and/or negative symptoms.

Clocapramine, also known as 3-chlorocarpipramine, is an atypical antipsychotic of the iminostilbene class which was introduced in Japan in 1974 by Yoshitomi for the treatment of schizophrenia. In addition to psychosis, clocapramine has also been used to augment antidepressants in the treatment of anxiety and panic.
Cariprazine, sold under the brand name Vraylar among others, is an atypical antipsychotic developed by Gedeon Richter, which is used in the treatment of schizophrenia, bipolar mania, bipolar depression, and major depressive disorder. It acts primarily as a D3 and D2 receptor partial agonist, with a preference for the D3 receptor. Cariprazine is also a partial agonist at the serotonin 5-HT1A receptor and acts as an antagonist at 5-HT2B and 5-HT2A receptors, with high selectivity for the D3 receptor. It is taken by mouth.
Panamesine (INNTooltip International Nonproprietary Name; developmental code name EMD-57455) is a sigma receptor antagonist that was under development by Merck as a potential antipsychotic for the treatment of schizophrenia in the 1990s but was never marketed. It is a selective antagonist of both sigma receptor subtypes, the σ1 and σ2 receptors (IC50 = 6 nM). In addition, the major metabolite of the drug, EMD-59983, has high affinity for the sigma receptors (IC50 = 24 nM) and the dopamine D2 (IC50 = 23 nM) and D3 receptors, with potent antidopaminergic activity. Panamesine reached phase II clinical trials for schizophrenia prior to the discontinuation of its development.


