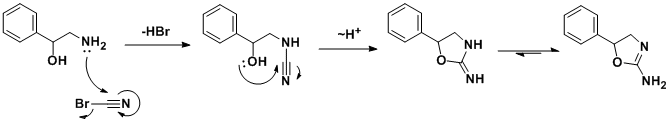Activation of serotonin 5-HT2B receptors by aminorex, either directly via agonism or indirectly via serotonin release, has been implicated in the development of pulmonary arterial hypertension and cardiac valvulopathy with the drug.[11][10][23][12] However, its EC50 for serotonin 5-HT2B receptor activation is 33-fold higher than its EC50 value for induction of norepinephrine release and is almost 50-fold less potent than the serotonin 5-HT2B receptor agonism of dexnorfenfluramine.[11] This seems to call into question the role of direct agonism of the serotonin 5-HT2B receptor in the toxicity of aminorex.[11] Along similar lines, chlorphentermine, a related drug that has also been associated with such adverse effects, shows negligible direct serotonin 5-HT2B receptor agonistic activity.[11] However, it is possible that metabolites of aminorex and chlorphentermine might be more potent in this action.[11]
The synthesis was first reported in a structure-activity relationship study of 2-amino-5-aryl-2-oxazolines, where aminorex was found to be approximately 2.5 times more potent than D-amphetamine sulfate in inducing anorexia in rats, and was also reported to have CNS stimulant effects.
The racemic synthesis involves addition/cyclization reaction of 2-amino-1-phenylethanol with cyanogen bromide.[30] A similar synthesis has been also published.[31] In a search for a cheaper synthetic route, a German team developed an alternative route[32] which, by using chiral styrene oxide, allows an enantiopure product.
History
It was discovered in 1962 by Edward John Hurlburt,[33] and was quickly found in 1963 to have an anorectic effect in rats. It was introduced as a prescription appetite suppressant in Germany, Switzerland and Austria in 1965, but was withdrawn in 1972 after it was found to cause pulmonary hypertension in approximately 0.2% of patients, and was linked to a number of deaths.[6][34]
↑Bertol E, Mari F, Milia MG, Politi L, Furlanetto S, Karch SB (July 2011). "Determination of aminorex in human urine samples by GC-MS after use of levamisole". Journal of Pharmaceutical and Biomedical Analysis. 55 (5): 1186–1189. doi:10.1016/j.jpba.2011.03.039. PMID21531521.
123456789Maier J, Mayer FP, Brandt SD, Sitte HH (October 2018). "DARK Classics in Chemical Neuroscience: Aminorex Analogues". ACS Chem Neurosci. 9 (10): 2484–2502. doi:10.1021/acschemneuro.8b00415. PMC6287711. PMID30269490. Due to the lack of interaction with the trace amine-associated receptor 1 (TAAR1), 4,4'- DMAR is suspected to be unable to trigger the auto-inhibitory pathway that, for example, MDMA possesses at least in rodents135,183,184. [...] As mentioned before, in contrast to other amphetamine-type stimulants, 4,4'-DMAR does not interact with TAAR1 and therefore lacks the auto-inhibitory pathway that attenuates monoamine release and mediates the neuroprotective effects231,232. It has however been shown that many psychoactive compounds stimulate human TAAR1 less potently than the receptor's rodent counterparts184.
123Partilla JS, Dersch CM, Baumann MH, Carroll FI, Rothman RB (1999). "Profiling CNS Stimulants with a High-Throughput Assay for Biogenic Amine Transporter Substrates". Problems of Drug Dependence 1999: Proceedings of the 61st Annual Scientific Meeting, The College on Problems of Drug Dependence, Inc(PDF). NIDA Res Monogr. Vol.180. pp.1–476 (252). PMID11680410. Archived from the original(PDF) on August 5, 2023. RESULTS. Methamphetamine and amphetamine potently released NE (IC50s = 14.3 and 7.0 nM) and DA (IC50s = 40.4 nM and 24.8 nM), and were much less potent releasers of 5-HT (IC50s = 740 nM and 1765 nM). Phentermine released all three biogenic amines with an order of potency NE (IC50 = 28.8 nM)> DA (IC50 = 262 nM)> 5-HT (IC50 = 2575 nM). Aminorex released NE (IC50 = 26.4 nM), DA (IC50 = 44.8 nM) and 5-HT (IC50 = 193 nM). Chlorphentermine was a very potent 5-HT releaser (IC50 = 18.2 nM), a weaker DA releaser (IC50 = 935 nM) and inactive in the NE release assay. Chlorphentermine was a moderate potency inhibitor of [3H]NE uptake (Ki = 451 nM). Diethylpropion, which is self-administered, was a weak DA uptake inhibitor (Ki = 15 µM) and NE uptake inhibitor (Ki = 18.1 µM) and essentially inactive in the other assays. Phendimetrazine, which is self-administered, was a weak DA uptake inhibitor (IC50 = 19 µM), a weak NE uptake inhibitor (8.3 µM) and essentially inactive in the other assays.
123Maier J, Mayer FP, Luethi D, Holy M, Jäntsch K, Reither H, etal. (August 2018). "The psychostimulant (±)-cis-4,4'-dimethylaminorex (4,4'-DMAR) interacts with human plasmalemmal and vesicular monoamine transporters". Neuropharmacology. 138: 282–291. doi:10.1016/j.neuropharm.2018.06.018. PMID29908239. Receptor-binding experiments suggest that 4,4'-DMAR exhibits no – or if at all only poor-affinity towards mouse and rat TAAR1. On the contrary, sub- (rat) and low-micromolar (mouse) affinities towards TAAR1 have been reported for MDMA (Simmler et al., 2013). The exact role of TAAR1 in amphetamine action remains far from being completely understood (Sitte and Freissmuth, 2015). However, TAAR1 appears to exert auto-inhibitory effects on monoaminergic neurons, thus regulates the release of the corresponding monoamines (Revel et al., 2011, 2012). TAAR1 is activated by a subset of amphetamines (Simmler et al., 2016). This observation has been linked to auto-inhibitory and neuroprotective effects of TAAR1 in amphetamine action (Miner et al., 2017; Revel et al., 2012; DiCara et al., 2011; Lindemann et al., 2008). The lack of agonist activity at TAAR1 might further contribute to long-term toxicity of 4,4'-DMAR, thus representing an interesting field for future investigations.
1234Rickli A, Kolaczynska K, Hoener MC, Liechti ME (May 2019). "Pharmacological characterization of the aminorex analogs 4-MAR, 4,4'-DMAR, and 3,4-DMAR". Neurotoxicology. 72: 95–100. Bibcode:2019NeuTx..72...95R. doi:10.1016/j.neuro.2019.02.011. PMID30776375. The methylated aminorex derivatives investigated in the present study did not interacted with TAAR1 receptors in contrast to amphetamine, MDMA, and several other phenethylamine derivatives (Revel et al., 2012; Simmler et al., 2016). Other aminorex-like ring-substituted 2- aminooxazolines have been shown to interact with TAAR1 receptors (Galley et al., 2016). However, they did not contain a 4-methyl group in contrast to the currently investigated compounds. Activity at TAAR1 may have auto-inhibitory effects on the monoaminergic action of amphetamine-type substances (Di Cara et al., 2011; Simmler et al., 2016). Therefore, the presently investigated compounds that did not bind to TAAR1 may exhibit greater stimulant properties compared to other amphetamines that also bind to TAAR1.
↑Ueda S, Terauchi H, Yano A, Ido M, Matsumoto M, Kawasaki M (January 2004). "4,5-Disubstituted-1,3-oxazolidin-2-imine derivatives: a new class of orally bioavailable nitric oxide synthase inhibitor". Bioorganic & Medicinal Chemistry Letters. 14 (2): 313–316. doi:10.1016/j.bmcl.2003.11.010. PMID14698148.
↑DE 2101424,"2-Amino-5-phenyl-2-oxazoline preparation", assigned to Polska Akademia Nauk Instytut Chemn Organicznej, Warschau
↑US 3115494,Albert MG, Ireland PG,"2-amino-5, 6-dihydro-4ii-1, 3-oxazines and a process for their preparation",issued 2 December 1963, assigned to Janssen Pharmaceuticals Inc.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.