
A receptor antagonist is a type of receptor ligand or drug that blocks or dampens a biological response by binding to and blocking a receptor rather than activating it like an agonist. Antagonist drugs interfere in the natural operation of receptor proteins. They are sometimes called blockers; examples include alpha blockers, beta blockers, and calcium channel blockers. In pharmacology, antagonists have affinity but no efficacy for their cognate receptors, and binding will disrupt the interaction and inhibit the function of an agonist or inverse agonist at receptors. Antagonists mediate their effects by binding to the active site or to the allosteric site on a receptor, or they may interact at unique binding sites not normally involved in the biological regulation of the receptor's activity. Antagonist activity may be reversible or irreversible depending on the longevity of the antagonist–receptor complex, which, in turn, depends on the nature of antagonist–receptor binding. The majority of drug antagonists achieve their potency by competing with endogenous ligands or substrates at structurally defined binding sites on receptors.

A dopamine antagonist, also known as an anti-dopaminergic and a dopamine receptor antagonist (DRA), is a type of drug which blocks dopamine receptors by receptor antagonism. Most antipsychotics are dopamine antagonists, and as such they have found use in treating schizophrenia, bipolar disorder, and stimulant psychosis. Several other dopamine antagonists are antiemetics used in the treatment of nausea and vomiting.
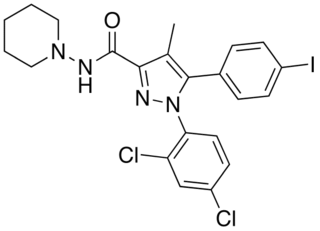
AM-251 is an inverse agonist at the CB1 cannabinoid receptor. AM-251 is structurally very close to rimonabant; both are biarylpyrazole cannabinoid receptor antagonists. In AM-251, the p-chloro group attached to the phenyl substituent at C-5 of the pyrazole ring is replaced with a p-iodo group. The resulting compound exhibits slightly better binding affinity for the CB1 receptor (with a Ki value of 7.5 nM) than rimonabant, which has a Ki value of 11.5 nM, AM-251 is, however, about two-fold more selective for the CB1 receptor when compared to rimonabant. Like rimonabant, it is additionally a μ-opioid receptor antagonist that attenuates analgesic effects.
The 5-HT2A receptor is a subtype of the 5-HT2 receptor that belongs to the serotonin receptor family and is a G protein-coupled receptor (GPCR). The 5-HT2A receptor is a cell surface receptor, but has several intracellular locations.
Neurokinin 1 (NK1) antagonists (-pitants) are a novel class of medications that possesses unique antidepressant, anxiolytic, and antiemetic properties. NK-1 antagonists boost the efficacy of 5-HT3 antagonists to prevent nausea and vomiting. The discovery of neurokinin 1 (NK1) receptor antagonists was a turning point in the prevention of nausea and vomiting associated with cancer chemotherapy.
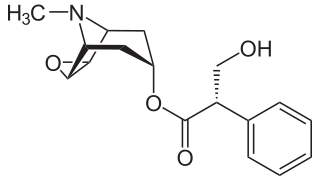
A muscarinic receptor antagonist (MRA), also called an antimuscarinic, is a type of anticholinergic agent that blocks the activity of the muscarinic acetylcholine receptor. The muscarinic receptor is a protein involved in the transmission of signals through certain parts of the nervous system, and muscarinic receptor antagonists work to prevent this transmission from occurring. Notably, muscarinic antagonists reduce the activation of the parasympathetic nervous system. The normal function of the parasympathetic system is often summarised as "rest-and-digest", and includes slowing of the heart, an increased rate of digestion, narrowing of the airways, promotion of urination, and sexual arousal. Muscarinic antagonists counter this parasympathetic "rest-and-digest" response, and also work elsewhere in both the central and peripheral nervous systems.

The 5-HT3 antagonists, informally known as "setrons", are a class of drugs that act as receptor antagonists at the 5-HT3 receptor, a subtype of serotonin receptor found in terminals of the vagus nerve and in certain areas of the brain. With the notable exceptions of alosetron and cilansetron, which are used in the treatment of irritable bowel syndrome, all 5-HT3 antagonists are antiemetics, used in the prevention and treatment of nausea and vomiting. They are particularly effective in controlling the nausea and vomiting produced by cancer chemotherapy and are considered the gold standard for this purpose.
A serotonin antagonist, or serotonin receptor antagonist, is a drug used to inhibit the action of serotonin and serotonergic drugs at serotonin (5-HT) receptors.

The adenosine A2B receptor, also known as ADORA2B, is a G-protein coupled adenosine receptor, and also denotes the human adenosine A2b receptor gene which encodes it.

The 5-HT7 receptor is a member of the GPCR superfamily of cell surface receptors and is activated by the neurotransmitter serotonin (5-hydroxytryptamine, 5-HT). The 5-HT7 receptor is coupled to Gs (stimulates the production of the intracellular signaling molecule cAMP) and is expressed in a variety of human tissues, particularly in the brain, the gastrointestinal tract, and in various blood vessels. This receptor has been a drug development target for the treatment of several clinical disorders. The 5-HT7 receptor is encoded by the HTR7 gene, which in humans is transcribed into 3 different splice variants.

Spiroxatrine is a drug which acts as a selective antagonist at both the 5-HT1A receptor and the α2C adrenergic receptor. It is an analog of spiperone and also has some dopamine antagonist effects.
A cannabinoid receptor antagonist, also known simply as a cannabinoid antagonist or as an anticannabinoid, is a type of cannabinoidergic drug that binds to cannabinoid receptors (CBR) and prevents their activation by endocannabinoids. They include antagonists, inverse agonists, and antibodies of CBRs. The discovery of the endocannabinoid system led to the development of CB1 receptor antagonists. The first CBR inverse agonist, rimonabant, was described in 1994. Rimonabant blocks the CB1 receptor selectively and has been shown to decrease food intake and regulate body-weight gain. The prevalence of obesity worldwide is increasing dramatically and has a great impact on public health. The lack of efficient and well-tolerated drugs to cure obesity has led to an increased interest in research and development of CBR antagonists. Cannabidiol (CBD), a naturally occurring cannabinoid and a non-competitive CB1/CB2 receptor antagonist, as well as Δ9-tetrahydrocannabivarin (THCV), a naturally occurring cannabinoid, modulate the effects of THC via direct blockade of cannabinoid CB1 receptors, thus behaving like first-generation CB1 receptor inverse agonists, such as rimonabant. CBD is a very low-affinity CB1 ligand, that can nevertheless affect CB1 receptor activity in vivo in an indirect manner, while THCV is a high-affinity CB1 receptor ligand and potent antagonist in vitro and yet only occasionally produces effects in vivo resulting from CB1 receptor antagonism. THCV has also high affinity for CB2 receptors and signals as a partial agonist, differing from both CBD and rimonabant.

Alazocine, also known more commonly as N-allylnormetazocine (NANM), is a synthetic opioid analgesic of the benzomorphan family related to metazocine, which was never marketed. In addition to its opioid activity, the drug is a sigma receptor agonist, and has been used widely in scientific research in studies of this receptor. Alazocine is described as a potent analgesic, psychotomimetic or hallucinogen, and opioid antagonist. Moreover, one of its enantiomers was the first compound that was found to selectively label the σ1 receptor, and led to the discovery and characterization of the receptor.
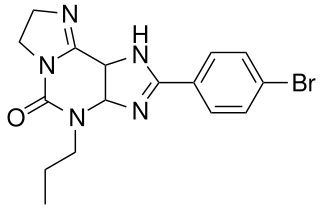
KF-26777 is a drug which acts as a potent and selective antagonist for the adenosine A3 receptor, with sub-nanomolar affinity (A3 Ki=0.2nM) and high selectivity over the other three adenosine receptor subtypes. Simple xanthine derivatives such as caffeine and DPCPX have generally low affinity for the A3 subtype and must be extended by expanding the ring system and adding an aromatic group to give high A3 affinity and selectivity.

Isamoltane (CGP-361A) is a drug used in scientific research. It acts as an antagonist at the β-adrenergic, 5-HT1A, and 5-HT1B receptors. It has about five times the potency for the 5-HT1B receptor over the 5-HT1A receptor. It has anxiolytic effects in rodents.
Serotonin antagonist and reuptake inhibitors (SARIs) are a class of drugs used mainly as antidepressants, but also as anxiolytics and hypnotics. They act by antagonizing serotonin receptors such as 5-HT2A and inhibiting the reuptake of serotonin, norepinephrine, and/or dopamine. Additionally, most also antagonize α1-adrenergic receptors. The majority of the currently marketed SARIs belong to the phenylpiperazine class of compounds.

Lomerizine (INN) is a diphenylpiperazine class L-type and T-type calcium channel blocker. This drug is currently used clinically for the treatment of migraines, while also being used experimentally for the treatment of glaucoma and optic nerve injury.

LY-2459989 is a silent antagonist of the κ-opioid receptor (KOR) that has been developed by Eli Lilly as a radiotracer of that receptor, labeled either with carbon-11 or fluorine-18. It possesses high affinity for the KOR and is highly selective for it over the μ-opioid receptor and the δ-opioid receptor. LY-2459989 is a fluorine-containing analogue and follow-up compound of LY-2795050, the first KOR-selective antagonist radiotracer. Relative to LY-2795050, LY-2459989 displays 4-fold higher affinity for the KOR and similar selectivity and also possesses greatly improved central nervous system permeation. The drug appears to possess a short duration of action, with only 25% remaining in serum at 30 minutes post-injection in rhesus monkeys, making it an ideal agent for application in biomedical imaging, for instance in positron emission tomography (PET).

2-Hydroxyestradiol (2-OHE2), also known as estra-1,3,5(10)-triene-2,3,17β-triol, is an endogenous steroid, catechol estrogen, and metabolite of estradiol, as well as a positional isomer of estriol.

Amesergide is a serotonin receptor antagonist of the ergoline and lysergamide families related to methysergide which was under development by Eli Lilly and Company for the treatment of a variety of conditions including depression, anxiety, schizophrenia, male sexual dysfunction, migraine, and thrombosis but was never marketed. It reached phase II clinical trials for the treatment of depression, erectile dysfunction, and premature ejaculation prior to the discontinuation of its development.
