
Phenethylamine (PEA) is an organic compound, natural monoamine alkaloid, and trace amine, which acts as a central nervous system stimulant in humans. In the brain, phenethylamine regulates monoamine neurotransmission by binding to trace amine-associated receptor 1 (TAAR1) and inhibiting vesicular monoamine transporter 2 (VMAT2) in monoamine neurons. To a lesser extent, it also acts as a neurotransmitter in the human central nervous system. In mammals, phenethylamine is produced from the amino acid L-phenylalanine by the enzyme aromatic L-amino acid decarboxylase via enzymatic decarboxylation. In addition to its presence in mammals, phenethylamine is found in many other organisms and foods, such as chocolate, especially after microbial fermentation.

Tryptamine is an indolamine metabolite of the essential amino acid tryptophan. The chemical structure is defined by an indole—a fused benzene and pyrrole ring, and a 2-aminoethyl group at the second carbon. The structure of tryptamine is a shared feature of certain aminergic neuromodulators including melatonin, serotonin, bufotenin and psychedelic derivatives such as dimethyltryptamine (DMT), psilocybin, psilocin and others.

3,4-Methylenedioxyamphetamine (MDA), sometimes referred to as sass, is an empathogen-entactogen, stimulant, and psychedelic drug of the amphetamine family that is encountered mainly as a recreational drug. In its pharmacology, MDA is a serotonin–norepinephrine–dopamine releasing agent (SNDRA). In most countries, the drug is a controlled substance and its possession and sale are illegal.
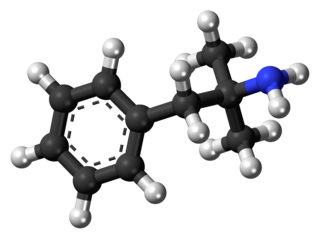
Phentermine, sold under the brand name Adipex-P among others, is a medication used together with diet and exercise to treat obesity. It is available by itself or as the combination phentermine/topiramate. Phentermine is taken by mouth.
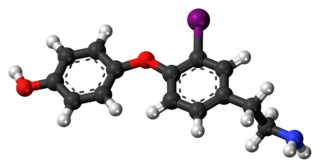
3-Iodothyronamine (T1AM) is an endogenous thyronamine. It is a high-affinity ligand of the trace amine-associated receptor 1 (TAAR1). T1AM is the most potent endogenous TAAR1 agonist yet discovered. It is also an agonist of the TAAR2 and TAAR5 with similar potency as for the TAAR1 (all in the case of the human proteins). T1AM is not a ligand of the thyroid hormone receptors. However, it is additionally a ligand of various monoamine and other receptors. For instance, it is a muscarinic acetylcholine receptor antagonist.

Trace amines are an endogenous group of trace amine-associated receptor 1 (TAAR1) agonists – and hence, monoaminergic neuromodulators – that are structurally and metabolically related to classical monoamine neurotransmitters. Compared to the classical monoamines, they are present in trace concentrations. They are distributed heterogeneously throughout the mammalian brain and peripheral nervous tissues and exhibit high rates of metabolism. Although they can be synthesized within parent monoamine neurotransmitter systems, there is evidence that suggests that some of them may comprise their own independent neurotransmitter systems.

Trace amine-associated receptor 1 (TAAR1) is a trace amine-associated receptor (TAAR) protein that in humans is encoded by the TAAR1 gene.
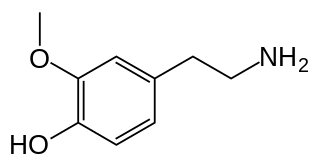
3-Methoxytyramine (3-MT), also known as 3-methoxy-4-hydroxyphenethylamine, is a human trace amine and the major metabolite of the monoamine neurotransmitter dopamine. It is formed by the introduction of a methyl group to dopamine by the enzyme catechol-O-methyltransferase (COMT). 3-MT can be further metabolized by the enzyme monoamine oxidase (MAO) to form homovanillic acid (HVA), which is then typically excreted in the urine.

para-Chloroamphetamine (PCA), also known as 4-chloroamphetamine (4-CA), is a serotonin–norepinephrine–dopamine releasing agent (SNDRA) and serotonergic neurotoxin of the amphetamine family. It is used in scientific research in the study of the serotonin system, as a serotonin releasing agent (SRA) at lower doses to produce serotonergic effects, and as a serotonergic neurotoxin at higher doses to produce long-lasting depletions of serotonin.
A monoamine releasing agent (MRA), or simply monoamine releaser, is a drug that induces the release of one or more monoamine neurotransmitters from the presynaptic neuron into the synapse, leading to an increase in the extracellular concentrations of the neurotransmitters and hence enhanced signaling by those neurotransmitters. The monoamine neurotransmitters include serotonin, norepinephrine, and dopamine; MRAs can induce the release of one or more of these neurotransmitters.

EPPTB, also known as RO5212773 or RO-5212773, is a drug developed by Hoffmann-La Roche which acts as a potent and selective antagonist or inverse agonist of the trace amine-associated receptor 1 (TAAR1). The drug was the first selective antagonist developed for the TAAR1. It is a potent agonist of the mouse and rat TAAR1, but is dramatically less potent as an agonist of the human TAAR1. EPPTB has been used in scientific research to demonstrate an important role for TAAR1 in regulation of dopaminergic signaling in the limbic system.

o-Phenyl-3-iodotyramine (o-PIT) is a drug which acts as a selective agonist for the trace amine-associated receptor 1 (TAAR1). It has reasonable selectivity for TAAR1 but relatively low potency, and is rapidly metabolised in vivo, making it less useful for research than newer ligands such as RO5166017. Its EC50Tooltip half-maximal effective concentration values have been reported to be 35 nM for the mouse TAAR1, 2.4 nM at the rat TAAR1, and 9.5 nM at the human TAAR1.

Locomotor activity is a measure of animal behavior which is employed in scientific research.
Monoaminergic activity enhancers (MAE), also known as catecholaminergic/serotonergic activity enhancers (CAE/SAE), are a class of drugs that enhance the action potential-evoked release of monoamine neurotransmitters in the nervous system. MAEs are distinct from monoamine releasing agents (MRAs) like amphetamine and fenfluramine in that they do not induce the release of monoamines from synaptic vesicles but rather potentiate only nerve impulse propagation-mediated monoamine release. That is, MAEs increase the amounts of monoamine neurotransmitters released by neurons per electrical impulse.

RO5256390 or RO-5256390 is a drug developed by Hoffmann-La Roche which acts as an agonist for the trace amine associated receptor 1 (TAAR1). It is a full agonist of the rat, cynomolgus monkey, and human TAAR1, but a partial agonist of the mouse TAAR1.

RTI-7470-44 is a potent and selective antagonist of the human trace amine-associated receptor 1 (TAAR1) which is used in scientific research. It was discovered in 2022 and is the first potent antagonist of the human TAAR1 to be identified, following the potent mouse TAAR1 inverse agonist EPPTB in 2009.

RO5263397, or RO-5263397, is a trace amine-associated receptor 1 (TAAR1) partial or full agonist which is used in scientific research. It is the most well-studied of all of the synthetic TAAR1 ligands. In addition to its use in research, RO5263397 is or was under development for potential clinical use as a medication.

RO5203648 is a trace amine-associated receptor 1 (TAAR1) partial agonist. It is a potent and highly selective partial agonist of both rodent and primate TAAR1. The drug suppresses the effects of psychostimulants like cocaine and methamphetamine. It also produces a variety of other behavioral effects, such as antidepressant-like, antipsychotic-like, and antiaddictive effects. Research with RO5203648 has led to interest in TAAR1 agonists for potential treatment of drug addiction. RO5203648 itself was not developed for potential medical use due to poor expected human pharmacokinetics.

RO5073012 is a selective low-efficacy partial agonist of the trace amine-associated receptor 1 (TAAR1) which has been used in scientific research. TAAR1 partial agonists like RO5073012 can have agonist- or antagonist-like effects at the TAAR1 depending on the context and level of TAAR1 signaling.

L-687,414 is a glycine-site NMDA receptor antagonist or low-efficacy partial agonist which is used in scientific research. It a close analogue of HA-966. The drug has been found to produce hyperlocomotion, analgesia or antinociceptive effects, anticonvulsant effects, and neuroprotective effects in animals. In contrast to uncompetitive NMDA receptor antagonists like ketamine and phencyclidine (PCP), L-687,414 has not been associated with the development of brain vacuoles in animals.
