Modelling biological systems is a significant task of systems biology and mathematical biology. Computational systems biology aims to develop and use efficient algorithms, data structures, visualization and communication tools with the goal of computer modelling of biological systems. It involves the use of computer simulations of biological systems, including cellular subsystems, to both analyze and visualize the complex connections of these cellular processes.
Metabolic network modelling, also known as metabolic network reconstruction or metabolic pathway analysis, allows for an in-depth insight into the molecular mechanisms of a particular organism. In particular, these models correlate the genome with molecular physiology. A reconstruction breaks down metabolic pathways into their respective reactions and enzymes, and analyzes them within the perspective of the entire network. In simplified terms, a reconstruction collects all of the relevant metabolic information of an organism and compiles it in a mathematical model. Validation and analysis of reconstructions can allow identification of key features of metabolism such as growth yield, resource distribution, network robustness, and gene essentiality. This knowledge can then be applied to create novel biotechnology.
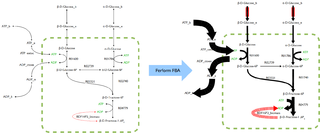
Flux balance analysis (FBA) is a mathematical method for simulating metabolism in genome-scale reconstructions of metabolic networks. In comparison to traditional methods of modeling, FBA is less intensive in terms of the input data required for constructing the model. Simulations performed using FBA are computationally inexpensive and can calculate steady-state metabolic fluxes for large models in a few seconds on modern personal computers. The related method of metabolic pathway analysis seeks to find and list all possible pathways between metabolites.
The Systems Biology Markup Language (SBML) is a representation format, based on XML, for communicating and storing computational models of biological processes. It is a free and open standard with widespread software support and a community of users and developers. SBML can represent many different classes of biological phenomena, including metabolic networks, cell signaling pathways, regulatory networks, infectious diseases, and many others. It has been proposed as a standard for representing computational models in systems biology today.

The Systems Biology Ontology (SBO) is a set of controlled, relational vocabularies of terms commonly used in systems biology, and in particular in computational modeling.
Systems immunology is a research field under systems biology that uses mathematical approaches and computational methods to examine the interactions within cellular and molecular networks of the immune system. The immune system has been thoroughly analyzed as regards to its components and function by using a "reductionist" approach, but its overall function can't be easily predicted by studying the characteristics of its isolated components because they strongly rely on the interactions among these numerous constituents. It focuses on in silico experiments rather than in vivo.
Fluxomics describes the various approaches that seek to determine the rates of metabolic reactions within a biological entity. While metabolomics can provide instantaneous information on the metabolites in a biological sample, metabolism is a dynamic process. The significance of fluxomics is that metabolic fluxes determine the cellular phenotype. It has the added advantage of being based on the metabolome which has fewer components than the genome or proteome.
Moiety conservation is the conservation of a subgroup in a chemical species, which is cyclically transferred from one molecule to another. In biochemistry, moiety conservation can have profound effects on the system's dynamics.
Physiomics is a systematic study of physiome in biology. Physiomics employs bioinformatics to construct networks of physiological features that are associated with genes, proteins and their networks. A few of the methods for determining individual relationships between the DNA sequence and physiological function include metabolic pathway engineering and RNAi analysis. The relationships derived from methods such as these are organized and processed computationally to form distinct networks. Computer models use these experimentally determined networks to develop further predictions of gene function.
Igor I. Goryanin is a systems biologist, who holds a Henrik Kacser Chair in Computational Systems Biology at the University of Edinburgh. He also heads the Biological Systems Unit at the Okinawa Institute of Science and Technology, Japan.
BioPAX is a RDF/OWL-based standard language to represent biological pathways at the molecular and cellular level. Its major use is to facilitate the exchange of pathway data. Pathway data captures our understanding of biological processes, but its rapid growth necessitates development of databases and computational tools to aid interpretation. However, the current fragmentation of pathway information across many databases with incompatible formats presents barriers to its effective use. BioPAX solves this problem by making pathway data substantially easier to collect, index, interpret and share. BioPAX can represent metabolic and signaling pathways, molecular and genetic interactions and gene regulation networks. BioPAX was created through a community process. Through BioPAX, millions of interactions organized into thousands of pathways across many organisms, from a growing number of sources, are available. Thus, large amounts of pathway data are available in a computable form to support visualization, analysis and biological discovery.
Process simulation is used for the design, development, analysis, and optimization of technical process of simulation of processes such as: chemical plant s, chemical processes, environmental systems, power stations, complex manufacturing operations, biological processes, and similar technical functions.

The minimum information about a simulation experiment (MIASE) is a list of the common set of information a modeller needs to enable the execution and reproduction of a numerical simulation experiment, derived from a given set of quantitative models.

Systrip is a visual environment for the analysis of time-series data in the context of biological networks. Systrip gathers bioinformatics and graph theoretical algorithms that can be assembled in different ways to help biologists in their visual mining process. It had been used to analyze various real biological data.
Virtual Cell (VCell) is an open-source software platform for modeling and simulation of living organisms, primarily cells. It has been designed to be a tool for a wide range of scientists, from experimental cell biologists to theoretical biophysicists.
LibSBML is an open-source software library that provides an application programming interface (API) for the SBML format. The libSBML library can be embedded in a software application or used in a web servlet as part of the application or servlet's implementation of support for reading, writing, and manipulating SBML documents and data streams. The core of libSBML is written in ISO standard C++; the library provides API for many programming languages via interfaces generated with the help of SWIG.
Multi-state modeling of biomolecules refers to a series of techniques used to represent and compute the behaviour of biological molecules or complexes that can adopt a large number of possible functional states.
Herbert M. Sauro works in the field of metabolic control analysis and systems biology.
libRoadRunner is a C/C++ software library that supports simulation of SBML based models.. It uses LLVM to generate extremely high-performance code and is the fastest SBML-based simulator currently available. Its main purpose is for use as a reusable library that can be hosted by other applications, particularly on large compute clusters for doing parameter optimization where performance is critical. It also has a set of Python bindings that allow it to be easily used from Python as well as a set of bindings for Julia.