In chemistry, a steady state is a situation in which all state variables are constant in spite of ongoing processes that strive to change them. For an entire system to be at steady state, i.e. for all state variables of a system to be constant, there must be a flow through the system (compare mass balance). A simple example of such a system is the case of a bathtub with the tap running but with the drain unplugged: after a certain time, the water flows in and out at the same rate, so the water level (the state variable Volume) stabilizes and the system is in a steady state.
The steady state concept is different from chemical equilibrium. Although both may create a situation where a concentration does not change, in a system at chemical equilibrium, the net reaction rate is zero (products transform into reactants at the same rate as reactants transform into products), while no such limitation exists in the steady state concept. Indeed, there does not have to be a reaction at all for a steady state to develop.
The term steady state is also used to describe a situation where some, but not all, of the state variables of a system are constant. For such a steady state to develop, the system does not have to be a flow system. Therefore, such a steady state can develop in a closed system where a series of chemical reactions take place. Literature in chemical kinetics usually refers to this case, calling it steady state approximation.
In simple systems the steady state is approached by state variables gradually decreasing or increasing until they reach their steady state value. In more complex systems state variables might fluctuate around the theoretical steady state either forever (a limit cycle) or gradually coming closer and closer. It theoretically takes an infinite time to reach steady state, just as it takes an infinite time to reach chemical equilibrium.
Both concepts are, however, frequently used approximations because of the substantial mathematical simplifications these concepts offer. Whether or not these concepts can be used depends on the error the underlying assumptions introduce. So, even though a steady state, from a theoretical point of view, requires constant drivers (e.g. constant inflow rate and constant concentrations in the inflow), the error introduced by assuming steady state for a system with non-constant drivers may be negligible if the steady state is approached fast enough (relatively speaking).
Steady state approximation in chemical kinetics
The steady state approximation,[1] occasionally called the stationary-state approximation or Bodenstein's quasi-steady state approximation, involves setting the rate of change of a reaction intermediate in a reaction mechanism equal to zero so that the kinetic equations can be simplified by setting the rate of formation of the intermediate equal to the rate of its destruction.
In practice it is sufficient that the rates of formation and destruction are approximately equal, which means that the net rate of variation of the concentration of the intermediate is small compared to the formation and destruction, and the concentration of the intermediate varies only slowly, similar to the reactants and products (see the equations and the green traces in the figures below).[citation needed]
As an example, the steady state approximation will be applied to two consecutive, irreversible, homogeneous first order reactions in a closed system. (For heterogeneous reactions, see reactions on surfaces.) This model corresponds, for example, to a series of nuclear decompositions like 239U → 239Np → 239Pu.
If the rate constants for the following reaction are k1 and k2; A → B → C, combining the rate equations with a mass balance for the system yields three coupled differential equations:
Reaction rates
For species A:
For species B:
Here the first (positive) term represents the formation of B by the first step A → B, whose rate depends on the initial reactant A. The second (negative) term represents the consumption of B by the second step B → C, whose rate depends on B as the reactant in that step.
For species C:
Analytical solutions
The analytical solutions for these equations (supposing that initial concentrations of every substance except for A are zero) are:[2]
Steady state
If the steady state approximation is applied, then the derivative of the concentration of the intermediate is set to zero. This reduces the second differential equation to an algebraic equation which is much easier to solve.
Therefore, so that
Since the concentration of the reaction intermediate B changes with the same time constant as [A] and is not in a steady state in that sense.
Validity
Concentration vs. time for k2/k1 = 0.5
Concentration of intermediate
Concentration of product
Concentration of substrate
Concentration vs. time for k2/k1 = 10
Concentration of intermediate
Concentration of product
Concentration of substrate
The analytical and approximated solutions should now be compared in order to decide when it is valid to use the steady state approximation. The analytical solution transforms into the approximate one when because then and Therefore, it is valid to apply the steady state approximation only if the second reaction is much faster than the first (k2/k1 > 10 is a common criterion), because that means that the intermediate forms slowly and reacts readily so its concentration stays low.
The graphs show concentrations of A (red), B (green) and C (blue) in two cases, calculated from the analytical solution.
When the first reaction is faster it is not valid to assume that the variation of [B] is very small, because [B] is neither low or close to constant: first A transforms into B rapidly and B accumulates because it disappears slowly. As the concentration of A decreases its rate of transformation decreases, at the same time the rate of reaction of B into C increases as more B is formed, so a maximum is reached when From then on the concentration of B decreases.
When the second reaction is faster, after a short induction period during which the steady state approximation does not apply, the concentration of B remains low (and more or less constant in an absolute sense) because its rates of formation and disappearance are almost equal and the steady state approximation can be used.
The equilibrium approximation can sometimes be used in chemical kinetics to yield similar results to the steady state approximation. It consists in assuming that the intermediate arrives rapidly at chemical equilibrium with the reactants. For example, Michaelis-Menten kinetics can be derived assuming equilibrium instead of steady state. Normally the requirements for applying the steady state approximation are laxer: the concentration of the intermediate is only needed to be low and more or less constant (as seen, this has to do only with the rates at which it appears and disappears) but it is not required to be at equilibrium.
Example
The reaction H2 + Br2 → 2 HBr has the following mechanism:
Br2 → 2Br
k1
Initiation
Br + H2 → HBr + H
k2
Propagation
H + Br2 → HBr + Br
k3
Propagation
H + HBr → H2 + Br
k4
Inhibition
2Br → Br2
k5
Breaking
The rate of each species are:
These equations cannot be solved, because each one has values that change with time. For example, the first equation contains the concentrations of [Br], [H2] and [Br2], which depend on time, as can be seen in their respective equations.
To solve the rate equations the steady state approximation can be used. The reactants of this reaction are H2 and Br2, the intermediates are H and Br, and the product is HBr.
For solving the equations, the rates of the intermediates are set to 0 in the steady state approximation:
From the reaction rate of H, k2[Br][H2] − k3[H][Br2] − k4[H][HBr] = 0 , so the reaction rate of Br can be simplified:
The reaction rate of HBr can also be simplifed, changing k2[Br][H2] − k4[H][Br] to k3[H][Br2], since both values are equal.
The concentration of H from equation 1 can be isolated:
The concentration of this intermediate is small and changes with time like the concentrations of reactants and product. It is inserted into the last differential equation to give
Simplifying the equation leads to
The experimentally observed rate is
The experimental rate law is the same as rate obtained with the steady state approximation, if is and is .
In a chemical reaction, chemical equilibrium is the state in which both the reactants and products are present in concentrations which have no further tendency to change with time, so that there is no observable change in the properties of the system. This state results when the forward reaction proceeds at the same rate as the reverse reaction. The reaction rates of the forward and backward reactions are generally not zero, but they are equal. Thus, there are no net changes in the concentrations of the reactants and products. Such a state is known as dynamic equilibrium.
A chain reaction is a sequence of reactions where a reactive product or by-product causes additional reactions to take place. In a chain reaction, positive feedback leads to a self-amplifying chain of events.
Half-life is the time required for a quantity to reduce to half of its initial value. The term is commonly used in nuclear physics to describe how quickly unstable atoms undergo radioactive decay or how long stable atoms survive. The term is also used more generally to characterize any type of exponential decay. For example, the medical sciences refer to the biological half-life of drugs and other chemicals in the human body. The converse of half-life is doubling time.
In chemistry, a dynamic equilibrium exists once a reversible reaction occurs. Substances initially transition between the reactants and products at different rates until the forward and backward reaction rates eventually equalize, meaning there is no net change. Reactants and products are formed at such a rate that the concentration of neither changes. It is a particular example of a system in a steady state.
The unimolecular nucleophilic substitution (SN1) reaction is a substitution reaction in organic chemistry. The Hughes-Ingold symbol of the mechanism expresses two properties—"SN" stands for "nucleophilic substitution", and the "1" says that the rate-determining step is unimolecular. Thus, the rate equation is often shown as having first-order dependence on the substrate and zero-order dependence on the nucleophile. This relationship holds for situations where the amount of nucleophile is much greater than that of the intermediate. Instead, the rate equation may be more accurately described using steady-state kinetics. The reaction involves a carbocation intermediate and is commonly seen in reactions of secondary or tertiary alkyl halides under strongly basic conditions or, under strongly acidic conditions, with secondary or tertiary alcohols. With primary and secondary alkyl halides, the alternative SN2 reaction occurs. In inorganic chemistry, the SN1 reaction is often known as the dissociative substitution. This dissociation pathway is well-described by the cis effect. A reaction mechanism was first introduced by Christopher Ingold et al. in 1940. This reaction does not depend much on the strength of the nucleophile, unlike the SN2 mechanism. This type of mechanism involves two steps. The first step is the ionization of alkyl halide in the presence of aqueous acetone or ethyl alcohol. This step provides a carbocation as an intermediate.
The reaction rate or rate of reaction is the speed at which a chemical reaction takes place, defined as proportional to the increase in the concentration of a product per unit time and to the decrease in the concentration of a reactant per unit time. Reaction rates can vary dramatically. For example, the oxidative rusting of iron under Earth's atmosphere is a slow reaction that can take many years, but the combustion of cellulose in a fire is a reaction that takes place in fractions of a second. For most reactions, the rate decreases as the reaction proceeds. A reaction's rate can be determined by measuring the changes in concentration over time.
In chemistry, the law of mass action is the proposition that the rate of a chemical reaction is directly proportional to the product of the activities or concentrations of the reactants. It explains and predicts behaviors of solutions in dynamic equilibrium. Specifically, it implies that for a chemical reaction mixture that is in equilibrium, the ratio between the concentration of reactants and products is constant.
Chemical kinetics, also known as reaction kinetics, is the branch of physical chemistry that is concerned with understanding the rates of chemical reactions. It is different from chemical thermodynamics, which deals with the direction in which a reaction occurs but in itself tells nothing about its rate. Chemical kinetics includes investigations of how experimental conditions influence the speed of a chemical reaction and yield information about the reaction's mechanism and transition states, as well as the construction of mathematical models that also can describe the characteristics of a chemical reaction.
In physical organic chemistry, a kinetic isotope effect (KIE) is the change in the reaction rate of a chemical reaction when one of the atoms in the reactants is replaced by one of its isotopes. Formally, it is the ratio of rate constants for the reactions involving the light (kL) and the heavy (kH) isotopically substituted reactants (isotopologues): KIE = kL/kH.
The equilibrium constant of a chemical reaction is the value of its reaction quotient at chemical equilibrium, a state approached by a dynamic chemical system after sufficient time has elapsed at which its composition has no measurable tendency towards further change. For a given set of reaction conditions, the equilibrium constant is independent of the initial analytical concentrations of the reactant and product species in the mixture. Thus, given the initial composition of a system, known equilibrium constant values can be used to determine the composition of the system at equilibrium. However, reaction parameters like temperature, solvent, and ionic strength may all influence the value of the equilibrium constant.
In chemical kinetics, the overall rate of a reaction is often approximately determined by the slowest step, known as the rate-determining step or rate-limiting step. For a given reaction mechanism, the prediction of the corresponding rate equation is often simplified by using this approximation of the rate-determining step.
In chemistry, the rate equation is an empirical differential mathematical expression for the reaction rate of a given reaction in terms of concentrations of chemical species and constant parameters only. For many reactions, the initial rate is given by a power law such as
In chemistry, molecularity is the number of molecules that come together to react in an elementary (single-step) reaction and is equal to the sum of stoichiometric coefficients of reactants in the elementary reaction with effective collision and correct orientation. Depending on how many molecules come together, a reaction can be unimolecular, bimolecular or even trimolecular.
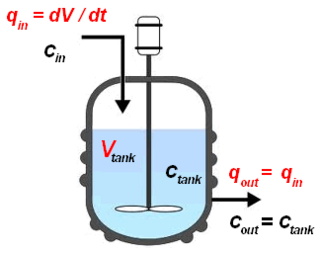
The continuous stirred-tank reactor (CSTR), also known as vat- or backmix reactor, mixed flow reactor (MFR), or a continuous-flow stirred-tank reactor (CFSTR), is a common model for a chemical reactor in chemical engineering and environmental engineering. A CSTR often refers to a model used to estimate the key unit operation variables when using a continuous agitated-tank reactor to reach a specified output. The mathematical model works for all fluids: liquids, gases, and slurries.
In physics, a mass balance, also called a material balance, is an application of conservation of mass to the analysis of physical systems. By accounting for material entering and leaving a system, mass flows can be identified which might have been unknown, or difficult to measure without this technique. The exact conservation law used in the analysis of the system depends on the context of the problem, but all revolve around mass conservation, i.e., that matter cannot disappear or be created spontaneously.
Reactions on surfaces are reactions in which at least one of the steps of the reaction mechanism is the adsorption of one or more reactants. The mechanisms for these reactions, and the rate equations are of extreme importance for heterogeneous catalysis. Via scanning tunneling microscopy, it is possible to observe reactions at the solid gas interface in real space, if the time scale of the reaction is in the correct range. Reactions at the solid–gas interface are in some cases related to catalysis.
An elementary reaction is a chemical reaction in which one or more chemical species react directly to form products in a single reaction step and with a single transition state. In practice, a reaction is assumed to be elementary if no reaction intermediates have been detected or need to be postulated to describe the reaction on a molecular scale. An apparently elementary reaction may be in fact a stepwise reaction, i.e. a complicated sequence of chemical reactions, with reaction intermediates of variable lifetimes.
In chemistry, transition state theory (TST) explains the reaction rates of elementary chemical reactions. The theory assumes a special type of chemical equilibrium (quasi-equilibrium) between reactants and activated transition state complexes.
In chemical kinetics, the Lindemann mechanism is a schematic reaction mechanism for unimolecular reactions. Frederick Lindemann and J.A. Christiansen proposed the concept almost simultaneously in 1921, and Cyril Hinshelwood developed it to take into account the energy distributed among vibrational degrees of freedom for some reaction steps.
Chemical reaction network theory is an area of applied mathematics that attempts to model the behaviour of real-world chemical systems. Since its foundation in the 1960s, it has attracted a growing research community, mainly due to its applications in biochemistry and theoretical chemistry. It has also attracted interest from pure mathematicians due to the interesting problems that arise from the mathematical structures involved.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.