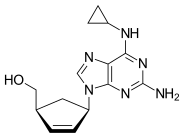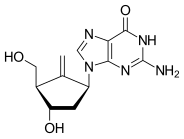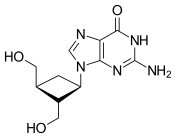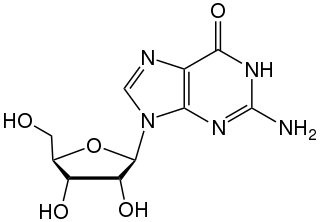
Guanosine (symbol G or Guo) is a purine nucleoside comprising guanine attached to a ribose (ribofuranose) ring via a β-N9-glycosidic bond. Guanosine can be phosphorylated to become guanosine monophosphate (GMP), cyclic guanosine monophosphate (cGMP), guanosine diphosphate (GDP), and guanosine triphosphate (GTP). These forms play important roles in various biochemical processes such as synthesis of nucleic acids and proteins, photosynthesis, muscle contraction, and intracellular signal transduction (cGMP). When guanine is attached by its N9 nitrogen to the C1 carbon of a deoxyribose ring it is known as deoxyguanosine.

Ribavirin, also known as tribavirin, is an antiviral medication used to treat RSV infection, hepatitis C and some viral hemorrhagic fevers. For hepatitis C, it is used in combination with other medications such as simeprevir, sofosbuvir, peginterferon alfa-2b or peginterferon alfa-2a. Among the viral hemorrhagic fevers it is sometimes used for Lassa fever, Crimean–Congo hemorrhagic fever, and Hantavirus infection but should not be used for Ebola or Marburg infections. Ribavirin is taken orally or inhaled. Despite widespread usage, since the 2010s it has faced scrutiny for a lack of efficacy in treating viral infections it has historically been prescribed for.

Aciclovir, also known as acyclovir, is an antiviral medication. It is primarily used for the treatment of herpes simplex virus infections, chickenpox, and shingles. Other uses include prevention of cytomegalovirus infections following transplant and severe complications of Epstein–Barr virus infection. It can be taken by mouth, applied as a cream, or injected.
Reverse-transcriptase inhibitors (RTIs) are a class of antiretroviral drugs used to treat HIV infection or AIDS, and in some cases hepatitis B. RTIs inhibit activity of reverse transcriptase, a viral DNA polymerase that is required for replication of HIV and other retroviruses.

Lamivudine, commonly called 3TC, is an antiretroviral medication used to prevent and treat HIV/AIDS. It is also used to treat chronic hepatitis B when other options are not possible. It is effective against both HIV-1 and HIV-2. It is typically used in combination with other antiretrovirals such as zidovudine, dolutegravir, and abacavir. Lamivudine may be included as part of post-exposure prevention in those who have been potentially exposed to HIV. Lamivudine is taken by mouth as a liquid or tablet.

Abacavir, sold under the brand name Ziagen among others, is a medication used to treat HIV/AIDS. Similar to other nucleoside analog reverse-transcriptase inhibitors (NRTIs), abacavir is used together with other HIV medications, and is not recommended by itself. It is taken by mouth as a tablet or solution and may be used in children over the age of three months.

Vidarabine or 9-β-D-arabinofuranosyladenine (ara-A) is an antiviral drug which is active against herpes simplex and varicella zoster viruses.

Entecavir (ETV), sold under the brand name Baraclude, is an antiviral medication used in the treatment of hepatitis B virus (HBV) infection. In those with both HIV/AIDS and HBV antiretroviral medication should also be used. Entecavir is taken by mouth as a tablet or solution.

Nucleoside analogues are structural analogues of a nucleoside, which normally contain a nucleobase and a sugar. Nucleotide analogues are analogues of a nucleotide, which normally has one to three phosphates linked to a nucleoside. Both types of compounds can deviate from what they mimick in a number of ways, as changes can be made to any of the constituent parts. They are related to nucleic acid analogues.
Non-nucleoside reverse-transcriptase inhibitors (NNRTIs) are antiretroviral drugs used in the treatment of human immunodeficiency virus (HIV). NNRTIs inhibit reverse transcriptase (RT), an enzyme that controls the replication of the genetic material of HIV. RT is one of the most popular targets in the field of antiretroviral drug development.
Discovery and development of nucleoside and nucleotide reverse-transcriptase inhibitors began in the 1980s when the AIDS epidemic hit Western societies. NRTIs inhibit the reverse transcriptase (RT), an enzyme that controls the replication of the genetic material of the human immunodeficiency virus (HIV). The first NRTI was zidovudine, approved by the U.S. Food and Drug Administration (FDA) in 1987, which was the first step towards treatment of HIV. Six NRTI agents and one NtRTI have followed. The NRTIs and the NtRTI are analogues of endogenous 2´-deoxy-nucleoside and nucleotide. Drug-resistant viruses are an inevitable consequence of prolonged exposure of HIV-1 to anti-HIV drugs.
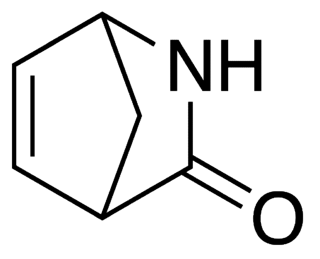
Vince lactam is the commercial name given to the bicyclic molecule γ-lactam 2-azabicyclo[2.2.1]hept-5-en-3-one. This lactam is a versatile chemical intermediate used in organic and medicinal chemistry. It is used as a synthetic precursor for three drugs. It is named after Robert Vince who has used the structural features of this molecule for the preparation of carbocyclic nucleosides. Vince's work with this lactam eventually led to his synthesis of abacavir. Peramivir synthesis is also dependent on Vince lactam starting material.
Topological inhibitors are rigid three-dimensional molecules of inorganic, organic, and hybrid compounds that form multicentered supramolecular interactions in vacant cavities of protein macromolecules and their complexes.

MK-608 is an antiviral drug, an adenosine analog. It was originally developed by Merck & Co. as a treatment for hepatitis C, but despite promising results in animal studies, it was ultimately unsuccessful in clinical trials. Subsequently it has been widely used in antiviral research and has shown activity against a range of viruses, including Dengue fever, tick-borne encephalitis virus, poliovirus, and most recently Zika virus, in both in vitro and animal models. Since it has already failed in human clinical trials previously, it is unlikely MK-608 itself will be developed as an antiviral medication, but the continuing lack of treatment options for these emerging viral diseases means that much research continues using MK-608 and related antiviral drugs.
HSV epigenetics is the epigenetic modification of herpes simplex virus (HSV) genetic code.
Non-structural protein 5B (NS5B) inhibitors are a class of direct-acting antivirals widely used in the treatment of chronic hepatitis C. Depending on site of action and chemical composition, NS5B inhibitors may be categorized into three classes—nucleoside active site inhibitors (NIs), non-nucleoside allosteric inhibitors, and pyrophosphate analogues. Subsequently, all three classes are then subclassified. All inhibit RNA synthesis by NS5B but at different stages/sites resulting in inability of viral RNA replication. Expression of direct-acting NS5B inhibitors does not take place in cells that are not infected by hepatitis C virus, which seems to be beneficial for this class of drugs.
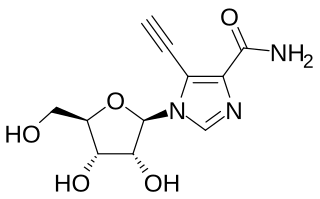
EICAR (5-Ethynyl-1-beta-D-ribofuranosylImidazole-4-CARboxamide) is a nucleoside analogue which has both anti-cancer and antiviral effects, and was originally developed for the treatment of leukemia, but was unsuccessful in human clinical trials. It has broad spectrum antiviral effects with activity against pox viruses, Semliki forest virus, Junin virus, reovirus, influenza, measles virus and respiratory syncytial virus among others, although it is not active against coronaviridae such as SARS-CoV-1. This useful spectrum of activity means that EICAR and related derivatives continue to be investigated for the treatment of viral diseases.

Azvudine is an antiviral drug which acts as a reverse transcriptase inhibitor. It was discovered for the treatment of hepatitis C and has since been investigated for use against other viral diseases such as AIDS and COVID-19, for which it was granted conditional approval in China.
Katherine Seley-Radtke is an American medicinal chemist who specializes in the discovery and design of novel nucleoside or nucleotide based enzyme inhibitors that may be used to treat infections or cancer. She has authored over 90 peer-reviewed publications, is an inventor of five issued US patents, and is a professor in the department of chemistry and biochemistry at the University of Maryland, Baltimore County. Her international impact includes scientific collaborations, policy advising and diplomatic appointments in biosecurity efforts.

Sangivamycin is a natural product originally isolated from Streptomyces rimosus, which is a nucleoside analogue. It acts as an inhibitor of protein kinase C. It has antibiotic, antiviral and anti-cancer properties and has been investigated for various medical applications, though never approved for clinical use itself. However, a number of related derivatives continue to be researched.