Related Research Articles

Molecular dynamics (MD) is a computer simulation method for analyzing the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanical force fields. The method is applied mostly in chemical physics, materials science, and biophysics.
In bioinformatics, sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of a wide range of analytical methods to understand its features, function, structure, or evolution. Methodologies used include sequence alignment, searches against biological databases, and others.
Chemistry at Harvard Macromolecular Mechanics (CHARMM) is the name of a widely used set of force fields for molecular dynamics, and the name for the molecular dynamics simulation and analysis computer software package associated with them. The CHARMM Development Project involves a worldwide network of developers working with Martin Karplus and his group at Harvard to develop and maintain the CHARMM program. Licenses for this software are available, for a fee, to people and groups working in academia.

Molecular mechanics uses classical mechanics to model molecular systems. The Born–Oppenheimer approximation is assumed valid and the potential energy of all systems is calculated as a function of the nuclear coordinates using force fields. Molecular mechanics can be used to study molecule systems ranging in size and complexity from small to large biological systems or material assemblies with many thousands to millions of atoms.

Biomolecular structure is the intricate folded, three-dimensional shape that is formed by a molecule of protein, DNA, or RNA, and that is important to its function. The structure of these molecules may be considered at any of several length scales ranging from the level of individual atoms to the relationships among entire protein subunits. This useful distinction among scales is often expressed as a decomposition of molecular structure into four levels: primary, secondary, tertiary, and quaternary. The scaffold for this multiscale organization of the molecule arises at the secondary level, where the fundamental structural elements are the molecule's various hydrogen bonds. This leads to several recognizable domains of protein structure and nucleic acid structure, including such secondary-structure features as alpha helixes and beta sheets for proteins, and hairpin loops, bulges, and internal loops for nucleic acids. The terms primary, secondary, tertiary, and quaternary structure were introduced by Kaj Ulrik Linderstrøm-Lang in his 1951 Lane Medical Lectures at Stanford University.
Nucleic acid structure prediction is a computational method to determine secondary and tertiary nucleic acid structure from its sequence. Secondary structure can be predicted from one or several nucleic acid sequences. Tertiary structure can be predicted from the sequence, or by comparative modeling.
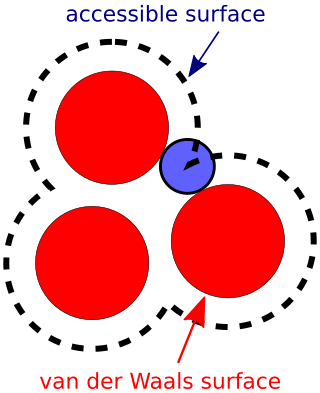
The accessible surface area (ASA) or solvent-accessible surface area (SASA) is the surface area of a biomolecule that is accessible to a solvent. Measurement of ASA is usually described in units of square angstroms. ASA was first described by Lee & Richards in 1971 and is sometimes called the Lee-Richards molecular surface. ASA is typically calculated using the 'rolling ball' algorithm developed by Shrake & Rupley in 1973. This algorithm uses a sphere of a particular radius to 'probe' the surface of the molecule.
This is a list of computer programs that are predominantly used for molecular mechanics calculations.
Nucleic acid methods are the techniques used to study nucleic acids: DNA and RNA.

Molecular modeling on GPU is the technique of using a graphics processing unit (GPU) for molecular simulations.

Molecular Dynamics of Mixtures (MDynaMix) is a computer software package for general purpose molecular dynamics to simulate mixtures of molecules, interacting by AMBER- and CHARMM-like force fields in periodic boundary conditions. Algorithms are included for NVE, NVT, NPT, anisotropic NPT ensembles, and Ewald summation to treat electrostatic interactions. The code was written in a mix of Fortran 77 and 90. The package runs on Unix and Unix-like (Linux) workstations, clusters of workstations, and on Windows in sequential mode.

Ascalaph Designer is a computer program for general purpose molecular modelling for molecular design and simulations. It provides a graphical environment for the common programs of quantum and classical molecular modelling ORCA, NWChem, Firefly, CP2K and MDynaMix . The molecular mechanics calculations cover model building, energy optimizations and molecular dynamics. Firefly covers a wide range of quantum chemistry methods. Ascalaph Designer is free and open-source software, released under the GNU General Public License, version 2 (GPLv2).
MacroModel is a computer program for molecular modelling of organic compounds and biopolymers. It features various chemistry force fields, plus energy minimizing algorithms, to predict geometry and relative conformational energies of molecules. MacroModel is maintained by Schrödinger, LLC.

Nucleic acid secondary structure is the basepairing interactions within a single nucleic acid polymer or between two polymers. It can be represented as a list of bases which are paired in a nucleic acid molecule. The secondary structures of biological DNAs and RNAs tend to be different: biological DNA mostly exists as fully base paired double helices, while biological RNA is single stranded and often forms complex and intricate base-pairing interactions due to its increased ability to form hydrogen bonds stemming from the extra hydroxyl group in the ribose sugar.
Martini is a coarse-grained (CG) force field developed by Marrink and coworkers at the University of Groningen, initially developed in 2004 for molecular dynamics simulation of lipids, later (2007) extended to various other molecules. The force field applies a mapping of four heavy atoms to one CG interaction site and is parametrized with the aim of reproducing thermodynamic properties.
Coarse-grained modeling, coarse-grained models, aim at simulating the behaviour of complex systems using their coarse-grained (simplified) representation. Coarse-grained models are widely used for molecular modeling of biomolecules at various granularity levels.
MBN Explorer is a software package for molecular dynamics simulations, structure optimization and kinetic Monte Carlo simulations. It is designed for multiscale computational analysis of structure and dynamics of atomic clusters and nanoparticles, biomolecules and nanosystems, nanostructured materials, different states of matter and various interfaces. The software has been developed by MBN Research Center.
Kenneth M. Merz Jr. is an American biochemist and molecular biologist currently the Joseph Zichis Chair and a distinguished university professor at Michigan State University and editor-in-chief of American Chemical Society's Journal of Chemical Information and Modeling. A highly cited expert in his field, his research interests are in computational chemistry and biology and computer-aided drug design (CADD). His group has been involved in developing the widely using AMBER suite of programs for simulating chemical and biological systems and the QUICK program for quantum chemical calculations.
References
- ↑ Cornell W.D.; Cieplak P.; Bayly C.I.; Gould I.R.; Merz K.M., Jr.; Ferguson D.M.; Spellmeyer D.C.; Fox T.; Caldwell J.W.; Kollman P.A. (1995). "A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules". J. Am. Chem. Soc. 117 (19): 5179–5197. CiteSeerX 10.1.1.323.4450 . doi:10.1021/ja00124a002.
- ↑ "The pmemd.cuda GPU Implementation".
- ↑ Wei, Wanlei; Luo, Jiaying; Waldispühl, Jérôme; Moitessier, Nicolas (24 June 2019). "Predicting Positions of Bridging Water Molecules in Nucleic Acid-Ligand Complexes". Journal of Chemical Information and Modeling. 59 (6): 2941–2951. doi:10.1021/acs.jcim.9b00163. ISSN 1549-960X. PMID 30998377. S2CID 121630416.
- ↑ Abagyan R.A., Totrov M.M. & Kuznetsov D.A. (1994). "Icm: A New Method For Protein Modeling and Design: Applications To Docking and Structure Prediction From The Distorted Native Conformation". J. Comput. Chem. 15 (5): 488–506. doi:10.1002/jcc.540150503. S2CID 206038130.
- ↑ Lavery, R., Zakrzewska, K. and Sklenar, H. (1995). "JUMNA: junction minimisation of nucleic acids". Comput. Phys. Commun. 91 (1–3): 135–158. Bibcode:1995CoPhC..91..135L. doi:10.1016/0010-4655(95)00046-I.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ A.P.Lyubartsev, A.Laaksonen (2000). "MDynaMix – A scalable portable parallel MD simulation package for arbitrary molecular mixtures". Computer Physics Communications. 128 (3): 565–589. Bibcode:2000CoPhC.128..565L. doi:10.1016/S0010-4655(99)00529-9.
- ↑ Macke T. & Case D.A. (1998). "Modeling unusual nucleic acid structures". Molecular Modeling of Nucleic Acids: 379–393.
- ↑ Petr Šulc; Flavio Romano; Thomas E. Ouldridge; Lorenzo Rovigatti; Jonathan P. K. Doye; Ard A. Louis (2012). "Sequence-dependent thermodynamics of a coarse-grained DNA model". J. Chem. Phys. 137 (13): 135101. arXiv: 1207.3391 . Bibcode:2012JChPh.137m5101S. doi:10.1063/1.4754132. PMID 23039613. S2CID 15555697.
- ↑ Oliver Henrich; Yair Augusto Gutiérrez Fosado; Tine Curk; Thomas E Ouldridge (2018). "Coarse-grained simulation of DNA using LAMMPS : An implementation of the oxDNA model and its applications". Eur. Phys. J. E. 41 (5): 57. arXiv: 1802.07145 . doi:10.1140/epje/i2018-11669-8. PMID 29748779. S2CID 3431325.
- ↑ Stasiewicz, Juliusz; Mukherjee, Sunandan; Nithin, Chandran; Bujnicki, Janusz M. (2019-03-21). "QRNAS: software tool for refinement of nucleic acid structures". BMC Structural Biology. 19 (1): 5. doi: 10.1186/s12900-019-0103-1 . ISSN 1472-6807. PMC 6429776 . PMID 30898165.
- ↑ Boniecki, Michal J.; Lach, Grzegorz; Dawson, Wayne K.; Tomala, Konrad; Lukasz, Pawel; Soltysinski, Tomasz; Rother, Kristian M.; Bujnicki, Janusz M. (2015-12-19). "SimRNA: a coarse-grained method for RNA folding simulations and 3D structure prediction". Nucleic Acids Research. 44 (7): e63. doi:10.1093/nar/gkv1479. ISSN 0305-1048. PMC 4838351 . PMID 26687716.
- ↑ Magnus, Marcin; Boniecki, Michał J.; Dawson, Wayne; Bujnicki, Janusz M. (2016-04-19). "SimRNAweb: a web server for RNA 3D structure modeling with optional restraints". Nucleic Acids Research. 44 (W1): W315–W319. doi:10.1093/nar/gkw279. ISSN 0305-1048. PMC 4987879 . PMID 27095203.