
Computational chemistry is a branch of chemistry that uses computer simulations to assist in solving chemical problems. It uses methods of theoretical chemistry incorporated into computer programs to calculate the structures and properties of molecules, groups of molecules, and solids. The importance of this subject stems from the fact that, with the exception of some relatively recent findings related to the hydrogen molecular ion, achieving an accurate quantum mechanical depiction of chemical systems analytically, or in a closed form, is not feasible. The complexity inherent in the many-body problem exacerbates the challenge of providing detailed descriptions of quantum mechanical systems. While computational results normally complement information obtained by chemical experiments, it can occasionally predict unobserved chemical phenomena.
Quantum chemistry, also called molecular quantum mechanics, is a branch of physical chemistry focused on the application of quantum mechanics to chemical systems, particularly towards the quantum-mechanical calculation of electronic contributions to physical and chemical properties of molecules, materials, and solutions at the atomic level. These calculations include systematically applied approximations intended to make calculations computationally feasible while still capturing as much information about important contributions to the computed wave functions as well as to observable properties such as structures, spectra, and thermodynamic properties. Quantum chemistry is also concerned with the computation of quantum effects on molecular dynamics and chemical kinetics.
Gaussian is a general purpose computational chemistry software package initially released in 1970 by John Pople and his research group at Carnegie Mellon University as Gaussian 70. It has been continuously updated since then. The name originates from Pople's use of Gaussian orbitals to speed up molecular electronic structure calculations as opposed to using Slater-type orbitals, a choice made to improve performance on the limited computing capacities of then-current computer hardware for Hartree–Fock calculations. The current version of the program is Gaussian 16. Originally available through the Quantum Chemistry Program Exchange, it was later licensed out of Carnegie Mellon University, and since 1987 has been developed and licensed by Gaussian, Inc.
Psi is an ab initio computational chemistry package originally written by the research group of Henry F. Schaefer, III. Utilizing Psi, one can perform a calculation on a molecular system with various kinds of methods such as Hartree-Fock, Post-Hartree–Fock electron correlation methods, and density functional theory. The program can compute energies, optimize molecular geometries, and compute vibrational frequencies. The major part of the program is written in C++, while Python API is also available, which allows users to perform complex computations or automate tasks easily.

In chemistry, pi stacking refers to the presumptive attractive, noncovalent pi interactions between the pi bonds of aromatic rings. However this is a misleading description of the phenomena since direct stacking of aromatic rings is electrostatically repulsive. What is more commonly observed is either a staggered stacking or pi-teeing interaction both of which are electrostatic attractive For example, the most commonly observed interactions between aromatic rings of amino acid residues in proteins is a staggered stacked followed by a perpendicular orientation. Sandwiched orientations are relatively rare.

Michael James Steuart Dewar was an American theoretical chemist.

Henry Frederick "Fritz" Schaefer III is a computational and theoretical chemist. He is one of the most highly cited chemists in the world, with a Thomson Reuters H-Index of 121 as of 2020. He is the Graham Perdue Professor of Chemistry and Director of the Center for Computational Chemistry at the University of Georgia. Before becoming professor at Georgia he was professor at University of California, Berkeley and in 2004, he became Professor of Chemistry Emeritus, at UC Berkeley

Spartan is a molecular modelling and computational chemistry application from Wavefunction. It contains code for molecular mechanics, semi-empirical methods, ab initio models, density functional models, post-Hartree–Fock models, and thermochemical recipes including G3(MP2) and T1. Quantum chemistry calculations in Spartan are powered by Q-Chem.
Semi-empirical quantum chemistry methods are based on the Hartree–Fock formalism, but make many approximations and obtain some parameters from empirical data. They are very important in computational chemistry for treating large molecules where the full Hartree–Fock method without the approximations is too expensive. The use of empirical parameters appears to allow some inclusion of electron correlation effects into the methods.

Cation–π interaction is a noncovalent molecular interaction between the face of an electron-rich π system (e.g. benzene, ethylene, acetylene) and an adjacent cation (e.g. Li+, Na+). This interaction is an example of noncovalent bonding between a monopole (cation) and a quadrupole (π system). Bonding energies are significant, with solution-phase values falling within the same order of magnitude as hydrogen bonds and salt bridges. Similar to these other non-covalent bonds, cation–π interactions play an important role in nature, particularly in protein structure, molecular recognition and enzyme catalysis. The effect has also been observed and put to use in synthetic systems.

Sir David Charles Clary, FRS is a British theoretical chemist. He was president of Magdalen College, Oxford, from 2005 to 2020. He was the first chief scientific adviser to the Foreign and Commonwealth Office from 2009 to 2013. He is a Professor of Chemistry at the University of Oxford.
Physical organic chemistry, a term coined by Louis Hammett in 1940, refers to a discipline of organic chemistry that focuses on the relationship between chemical structures and reactivity, in particular, applying experimental tools of physical chemistry to the study of organic molecules. Specific focal points of study include the rates of organic reactions, the relative chemical stabilities of the starting materials, reactive intermediates, transition states, and products of chemical reactions, and non-covalent aspects of solvation and molecular interactions that influence chemical reactivity. Such studies provide theoretical and practical frameworks to understand how changes in structure in solution or solid-state contexts impact reaction mechanism and rate for each organic reaction of interest.

Donald Gene Truhlar is an American scientist working in theoretical and computational chemistry and chemical physics with special emphases on quantum mechanics and chemical dynamics.
Robert J. Harrison is a distinguished expert in high-performance computing. He is a professor in the Applied Mathematics and Statistics department and founding Director of the Institute for Advanced Computational Science at Stony Brook University with a $20M endowment. Through a joint appointment with Brookhaven National Laboratory, Professor Harrison has also been named Director of the Computational Science Center and New York Center for Computational Sciences at Brookhaven. Dr. Harrison comes to Stony Brook from the University of Tennessee and Oak Ridge National Laboratory, where he was Director of the Joint Institute of Computational Science, Professor of Chemistry and Corporate Fellow. He has a prolific career in high-performance computing with over one hundred publications on the subject, as well as extensive service on national advisory committees.

Gregory A. Voth is a theoretical chemist and Haig P. Papazian Distinguished Service Professor of Chemistry at the University of Chicago. He is also a professor of the James Franck Institute and the Institute for Biophysical Dynamics.

Marcin Maciej Hoffmann is a Polish scientist and entrepreneur. He is a professor of chemistry at the Faculty of Chemistry of Adam Mickiewicz University in Poznań.

Hans Lischka is an Austrian computational theoretical chemist specialized on development and application of multireference methods for the study of molecular excited states. He is the main developer of the software package Columbus for ab initio multireference calculations and co-developer of the Newton-X program.

Laura Gagliardi is an Italian theoretical and computational chemist and the Richard and Kathy Leventhal Professor of Chemistry and Molecular Engineering at the University of Chicago. She is known for her work on the development of electronic structure methods and their use for understanding complex chemical systems.
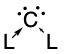
Carbones are a class of molecules containing a carbon atom in the 1D excited state with a formal oxidation state of zero where all four valence electrons exist as unbonded lone pairs. These carbon-based compounds are of the formula CL2 where L is a strongly σ-donating ligand, typically a phosphine (carbodiphosphoranes) or a N-heterocyclic carbene/NHC (carbodicarbenes), that stabilises the central carbon atom through donor-acceptor bonds. Carbones possess high-energy orbitals with both σ- and π-symmetry, making them strong Lewis bases and strong π-backdonor substituents. Carbones possess high proton affinities and are strong nucleophiles which allows them to function as ligands in a variety of main group and transition metal complexes. Carbone-coordinated elements also exhibit a variety of different reactivities and catalyse various organic and main group reactions.

Keiji Morokuma was a Japanese theoretical chemist and chemical engineer known for developing energy decomposition analysis for molecular interactions and the ONIOM method in quantum chemistry.
