
The polymerase chain reaction (PCR) is a method widely used to make millions to billions of copies of a specific DNA sample rapidly, allowing scientists to amplify a very small sample of DNA sufficiently to enable detailed study. PCR was invented in 1983 by American biochemist Kary Mullis at Cetus Corporation. Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.

A primer is a short single-stranded nucleic acid used by all living organisms in the initiation of DNA synthesis. A synthetic primer may also be referred to as an oligo, short for oligonucleotide. DNA polymerase enzymes are only capable of adding nucleotides to the 3’-end of an existing nucleic acid, requiring a primer be bound to the template before DNA polymerase can begin a complementary strand. DNA polymerase adds nucleotides after binding to the RNA primer and synthesizes the whole strand. Later, the RNA strands must be removed accurately and replace them with DNA nucleotides forming a gap region known as a nick that is filled in using an enzyme called ligase. The removal process of the RNA primer requires several enzymes, such as Fen1, Lig1, and others that work in coordination with DNA polymerase, to ensure the removal of the RNA nucleotides and the addition of DNA nucleotides. Living organisms use solely RNA primers, while laboratory techniques in biochemistry and molecular biology that require in vitro DNA synthesis usually use DNA primers, since they are more temperature stable. Primers can be designed in laboratory for specific reactions such as polymerase chain reaction (PCR). When designing PCR primers, there are specific measures that must be taken into consideration, like the melting temperature of the primers and the annealing temperature of the reaction itself. Moreover, the DNA binding sequence of the primer in vitro has to be specifically chosen, which is done using a method called basic local alignment search tool (BLAST) that scans the DNA and finds specific and unique regions for the primer to bind.
In genetics and biochemistry, sequencing means to determine the primary structure of an unbranched biopolymer. Sequencing results in a symbolic linear depiction known as a sequence which succinctly summarizes much of the atomic-level structure of the sequenced molecule.

Genomics is an interdisciplinary field of biology focusing on the structure, function, evolution, mapping, and editing of genomes. A genome is an organism's complete set of DNA, including all of its genes as well as its hierarchical, three-dimensional structural configuration. In contrast to genetics, which refers to the study of individual genes and their roles in inheritance, genomics aims at the collective characterization and quantification of all of an organism's genes, their interrelations and influence on the organism. Genes may direct the production of proteins with the assistance of enzymes and messenger molecules. In turn, proteins make up body structures such as organs and tissues as well as control chemical reactions and carry signals between cells. Genomics also involves the sequencing and analysis of genomes through uses of high throughput DNA sequencing and bioinformatics to assemble and analyze the function and structure of entire genomes. Advances in genomics have triggered a revolution in discovery-based research and systems biology to facilitate understanding of even the most complex biological systems such as the brain.

Frederick Sanger was a British biochemist who received the Nobel Prize in Chemistry twice.

DNA polymerase I is an enzyme that participates in the process of prokaryotic DNA replication. Discovered by Arthur Kornberg in 1956, it was the first known DNA polymerase. It was initially characterized in E. coli and is ubiquitous in prokaryotes. In E. coli and many other bacteria, the gene that encodes Pol I is known as polA. The E. coli Pol I enzyme is composed of 928 amino acids, and is an example of a processive enzyme — it can sequentially catalyze multiple polymerisation steps without releasing the single-stranded template. The physiological function of Pol I is mainly to support repair of damaged DNA, but it also contributes to connecting Okazaki fragments by deleting RNA primers and replacing the ribonucleotides with DNA.
Pyrosequencing is a method of DNA sequencing based on the "sequencing by synthesis" principle, in which the sequencing is performed by detecting the nucleotide incorporated by a DNA polymerase. Pyrosequencing relies on light detection based on a chain reaction when pyrophosphate is released. Hence, the name pyrosequencing.
DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.

Sanger sequencing is a method of DNA sequencing that involves electrophoresis and is based on the random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. After first being developed by Frederick Sanger and colleagues in 1977, it became the most widely used sequencing method for approximately 40 years. It was first commercialized by Applied Biosystems in 1986. More recently, higher volume Sanger sequencing has been replaced by next generation sequencing methods, especially for large-scale, automated genome analyses. However, the Sanger method remains in wide use for smaller-scale projects and for validation of deep sequencing results. It still has the advantage over short-read sequencing technologies in that it can produce DNA sequence reads of > 500 nucleotides and maintains a very low error rate with accuracies around 99.99%. Sanger sequencing is still actively being used in efforts for public health initiatives such as sequencing the spike protein from SARS-CoV-2 as well as for the surveillance of norovirus outbreaks through the Center for Disease Control and Prevention's (CDC) CaliciNet surveillance network.
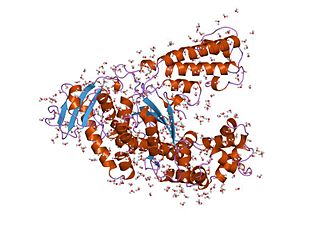
Taq polymerase is a thermostable DNA polymerase I named after the thermophilic eubacterial microorganism Thermus aquaticus, from which it was originally isolated by Chien et al. in 1976. Its name is often abbreviated to Taq or Taq pol. It is frequently used in the polymerase chain reaction (PCR), a method for greatly amplifying the quantity of short segments of DNA.
Allan Maxam is one of the pioneers of molecular genetics. He was one of the contributors to develop a DNA sequencing method at Harvard University, while working as a student in the laboratory of Walter Gilbert.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.
The polymerase chain reaction (PCR) is a commonly used molecular biology tool for amplifying DNA, and various techniques for PCR optimization which have been developed by molecular biologists to improve PCR performance and minimize failure.

The history of the polymerase chain reaction (PCR) has variously been described as a classic "Eureka!" moment, or as an example of cooperative teamwork between disparate researchers. Following is a list of events before, during, and after its development:

T7 DNA polymerase is an enzyme used during the DNA replication of the T7 bacteriophage. During this process, the DNA polymerase “reads” existing DNA strands and creates two new strands that match the existing ones. The T7 DNA polymerase requires a host factor, E. coli thioredoxin, in order to carry out its function. This helps stabilize the binding of the necessary protein to the primer-template to improve processivity by more than 100-fold, which is a feature unique to this enzyme. It is a member of the Family A DNA polymerases, which include E. coli DNA polymerase I and Taq DNA polymerase.

Ion semiconductor sequencing is a method of DNA sequencing based on the detection of hydrogen ions that are released during the polymerization of DNA. This is a method of "sequencing by synthesis", during which a complementary strand is built based on the sequence of a template strand.
Massive parallel sequencing or massively parallel sequencing is any of several high-throughput approaches to DNA sequencing using the concept of massively parallel processing; it is also called next-generation sequencing (NGS) or second-generation sequencing. Some of these technologies emerged between 1993 and 1998 and have been commercially available since 2005. These technologies use miniaturized and parallelized platforms for sequencing of 1 million to 43 billion short reads per instrument run.

Illumina dye sequencing is a technique used to determine the series of base pairs in DNA, also known as DNA sequencing. The reversible terminated chemistry concept was invented by Bruno Canard and Simon Sarfati at the Pasteur Institute in Paris. It was developed by Shankar Balasubramanian and David Klenerman of Cambridge University, who subsequently founded Solexa, a company later acquired by Illumina. This sequencing method is based on reversible dye-terminators that enable the identification of single nucleotides as they are washed over DNA strands. It can also be used for whole-genome and region sequencing, transcriptome analysis, metagenomics, small RNA discovery, methylation profiling, and genome-wide protein-nucleic acid interaction analysis.

Maxam–Gilbert sequencing is a method of DNA sequencing developed by Allan Maxam and Walter Gilbert in 1976–1977. This method is based on nucleobase-specific partial chemical modification of DNA and subsequent cleavage of the DNA backbone at sites adjacent to the modified nucleotides.
In molecular biology, hybridization is a phenomenon in which single-stranded deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) molecules anneal to complementary DNA or RNA. Though a double-stranded DNA sequence is generally stable under physiological conditions, changing these conditions in the laboratory will cause the molecules to separate into single strands. These strands are complementary to each other but may also be complementary to other sequences present in their surroundings. Lowering the surrounding temperature allows the single-stranded molecules to anneal or “hybridize” to each other.

