The epigenetics of schizophrenia is the study of how inherited epigenetic changes are regulated and modified by the environment and external factors and how these changes influence the onset and development of, and vulnerability to, schizophrenia. Epigenetics concerns the heritability of those changes, too. Schizophrenia is a debilitating and often misunderstood disorder that affects up to 1% of the world's population.[1] Although schizophrenia is a heavily studied disorder, it has remained largely impervious to scientific understanding; epigenetics offers a new avenue for research, understanding, and treatment.
Historically, schizophrenia has been studied and examined through different paradigms, or schools of thought. In the late 1870s, Emil Kraepelin started the idea of studying it as an illness. Another paradigm, introduced by Zubin and Spring in 1977, was the stress-vulnerability model where the individual has unique characteristics that give him or her strengths or vulnerabilities to deal with stress, a predisposition for schizophrenia. More recently, with the decoding of the human genome, there had been a focus on identifying specific genes to study the disease. However, the genetics paradigm faced problems with inconsistent, inconclusive, and variable results. The most recent school of thought is studying schizophrenia through epigenetics.[2]
A visualization of the stress-vulnerability model, also known as the diathesis-stress model
The idea of epigenetics has been described as far back as 1942, when Conrad Waddington described it as how the environment regulated genetics. As the field and available technology has progressed, the term has come to also refer to the molecular mechanisms of regulation. The concept that these epigenetic changes can be passed on to future generations has progressively become more accepted.[3]
While epigenetics is a relatively new field of study, specific applications and focus on mental disorders like schizophrenia is an even more recent area of research.
Schizophrenia
Symptoms
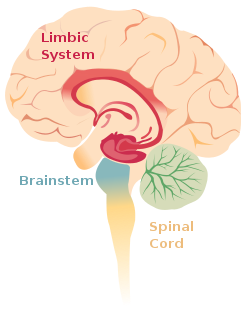
The core symptoms of schizophrenia can be classified into three broad categories. These symptoms are often used to build and study animal models of schizophrenia in the field of epigenetics.[1] Positive symptoms are considered limbic system aberrations, while negative and cognitive symptoms are thought of as frontal lobe abnormalities.[4]
Positive symptoms:
The limbic system and associated structures of the brain, where abnormalities lead to positive symptoms of schizophrenia
Heritability represents the percentage in variability in a trait that comes from genetic differences. There is a great deal of evidence to show that schizophrenia is a heritable disease. It has been calculated that the heritability of schizophrenia is around 80%, and within that heritability 30% of the variability comes from single nucleotide polymorphisms and another 30% from large copy number variants (CNVs). One key piece of evidence is a twin study that showed that the likelihood of developing the disease is 53% for one member of monozygotic twins (twins with same genetic code), compared to the 15% for dizygotic twins, who don't share the exact DNA.[5] It has also been shown that having direct family members that are diagnosed with schizophrenia increases the rate of developing schizophrenia by 9 times. Another study investigated the rates of schizophrenia of adoptees: adoptees with a genetic history of schizophrenia that were adopted by non-schizophrenic parents were seen to be diagnosed with schizophrenia at higher rates than adoptees without a biological history of schizophrenia but raised with adoptive parents diagnosed with schizophrenia.[1] However, the extent to which interactions between environmental and genetic factors contribute to the development of schizophrenia is uncertain. Others question the evidence of heritability due to different definitions of schizophrenia and the similar environment for both twins.[6][7] That said, the fact that even monozygotic twins don't share a 100% concordance rate suggests environmental factors play a role in the vulnerability and development of the disorder.
Maternal immune responses are involved in schizophrenia development. A 2019 study found that, in mice models, maternal immune activation may be involved in the regulation of the ARX gene expression that contributes to the GABA dysfunction commonly implicated in schizophrenia.[8] Most maternal effects involved in schizophrenia development have to do with immune system activation and associated cytokine and neurogenesis mechanisms rather than genetic changes.
Paternal genetic effects are seen to be involved in the rate of development of schizophrenia. A 2021 study showed that advanced paternal age was associated with higher schizophrenia risk. In the study, the paternal ages were grouped into 5-year increments, and it was seen that as the increments increased, so did the risk of developing schizophrenia.[9] It is hypothesized that advanced paternal age increases schizophrenia risk because fathers pass down three to four times more de novo mutations than do mothers.[10] It is estimated that 2 new de novo mutations are created a year, resulting in fathers with an advanced age passing down more mutations to their offspring, which may explain the increased rates of schizophrenia.[10]
There are various environmental factors that have been suggested, including the use of marijuana, complications during pregnancy, socioeconomic status and environment, and maternal malnutrition. In some circumstances, it has been shown that environmental factors can increase the risk of schizophrenia when combined with a family history of psychosis.[11] As the field of epigenetics advances, these and other external risk factors are likely to be considered in epidemiological studies.[1]
Genes associated with schizophrenia
Among the many genes known to be associated with schizophrenia, a few have been identified as particularly important when studying the epigenetic phenomena underlying the disease.
GAD1 – GAD1 codes for the proteinGAD67, an enzyme that catalyzes the formation of GABA from glutamate. Individuals with schizophrenia have shown a decrease in GAD67 levels and this deficit is thought to lead to working memory problems, among other impairments.[12]
RELN – RELN codes for reelin, an extracellular protein that is necessary for formation of memories and learning through plasticity. Reelin is thought to regulate nearby glutamate producing neurons.[1]
Both proteins are expressed by GABAergic neurons. Several studies have demonstrated that levels of both reelin and GAD67 are downregulated in patients with schizophrenia and animal models.
BDNF – Brain-derived neurotrophic factor, BDNF, is another important gene in the study of schizophrenia genetics. BDNF plays a crucial role in cognition, learning, memory formation, and vulnerability to social and life experiences.[1]
Genome-wide association studies (GWAS) have confirmed the presence of other genes thought to be involved in the regulation of schizophrenia.[11] Several genes that GWAS have determined to be associated with schizophrenia include genes involved in neurodevelopment, including glutamatergic signaling, synaptic transmission, and the regulation of voltage-gated calcium channels.[13] This includes the GAD1 and RELN pathways denoted above as well as BDNF. Another important gene is the Dopamine Receptor 2 gene, which encodes a dopamine receptor (D2), a primary target of many of the antipsychotic drugs that are used to treat patients with schizophrenia.[13][14] Chromosomal regions containing a large number of CNVs are reported to lead to increased susceptibility to schizophrenia.[11]
Copy number variants (CNVs) have also been reported to be associated with schizophrenia, particularly in chromosomal regions with large amounts of CNVs.[13][11] A CNV located at the gene NRXN1, which encodes a neurexin protein involved in synaptic transmission, is thought to cause a loss-of-function mutation that is associated with the development of schizophrenia.[13] Loss-of-function mutations at the gene encoding histone H3 methyltransferase, an important enzyme for epigenetic histone modification, have also been implicated in gene-association studies for schizophrenia.[13] Histone modification is not the only epigenetic mechanism thought to be commonly associated with schizophrenia. Analyzing schizophrenia-associated genes reveals that risk loci are commonly found near DNA methylation quantitative trait loci (which affect CpG methylation), which have post-transcriptional modifications uniquely correlated to schizophrenia and produce splice variants with strong chromatin associations.[13] DNA methylation, post-transcriptional modification and splice variation, and chromatin modifications are all prominent epigenetic mechanisms, and their association with schizophrenia-risk-associated loci indicates that these mechanisms may play a significant role in the development of the disease.
Moving away from GWAS, linkage studies have proved to be unsuccessful due to the interaction of several different genes that are all involved in the development of schizophrenia.[11] There are also few specific SNPs majorly involved in the development of schizophrenia, however groups of SNPs could account for 30% of the genetic susceptibility for developing schizophrenia.[13][11]
Research methods
Epigenetics can be studied and researched through various methods. One of the most common methods is looking at postmortem brain tissue of patients with schizophrenia and analyzing them for biomarkers. Other common methods include tissue culture studies of neurons, genome-wide analysis of non-brain cells in living patients (see PBMC), and transgenic and schizophrenic animal models.[1]
Other studies that are currently being done or that can be done in the future include longitudinal studies of patients, "at-risk" populations, and monozygotic twins, and studies that examine specific gene-environment interactions and epigenetic effects.[15]
Epigenetic alterations
Epigenetics (translated as "above genetics") is the study of how genes are regulated through reversible and heritable molecular mechanisms. The epigenetic changes modify gene expression through either activation of the gene that codes for a certain protein, or repression of the gene. There are two main categories of modifications: the methylation of DNA and modifications to histones. Research findings have demonstrated that several examples of both of these changes are linked to schizophrenia and its symptoms.[13][1] Evidence of epigenetic regulation is still sparse, and just as genetic culprits for the disease remain nebulous, there is no definite answer to what epigenetic alterations should be expected in patients with schizophrenia.
Mechanisms of epigenetics in the cell
DNA methylation
DNA methylation is the covalent addition of a methyl group to a segment of the DNA code. These -CH3 groups are added to cytosine residues by the DNMT (DNA Methytransferases) enzymes. The binding methyl group to promoter regions interferes with the binding of transcription factors and silences the gene by preventing the transcription of that code.[1] DNA methylation is one of the most well studied epigenetic mechanisms and there have been several findings linking it to schizophrenia.
Differential DNA methylation has been identified in the schizophrenic epigenome across 4 different regions of the brain.[13] Hypermethylation of genes in neurotransmitter pathways (including GAD1, RELN, and the serotonin pathway) as well as hypomethylation of genes in other pathways (such as the dopaminergic pathway) have been observed in schizophrenic patients.[14][13] A broad range of genomic methylation patterns have been observed in patients with schizophrenia, and although a definite explanation is not in place, there are enough consistent abnormalities to suspect that differential methylation may play a substantial role in the pathogenesis of schizophrenia.
Methylation of GABAergic genes
It has been consistently shown in various studies that levels of reelin and GAD67 are downregulated in the cortical and hippocampal tissue samples of individuals with schizophrenia. These proteins are used by GABAergic neurons, and abnormalities in their levels could result in some of the symptoms found in individuals with schizophrenia. The genes for these two proteins are found in areas of the genetic code that can be methylated (see CpG island). Recent studies have demonstrated an epigenetic link between the levels of the proteins and schizophrenia. One study found that cortical neurons with lower levels of GAD67 and reelin also showed increased levels of DNMT1, one of the enzymes that adds a methyl group. It has also been shown that a schizophrenic-type state can be induced in mice when they were chronically given l-methionine, a precursor necessary for DNMT activity. These and other findings provide a strong link between epigenetic changes and schizophrenia.[12]
Methylation of BDNF
DNA methylation can also affect expression of BDNF (brain derived neurotrophic factor). The BDNF protein is important for cognition, learning, and even vulnerability to early life trauma. Sun et al. showed that fear condition led to changes in DNA methylation levels in BDNF promoter regions in hippocampal neurons. It was also shown that inhibiting DNMT activity led to change in levels of BDNF in the hippocampus. Methylation of BDNF DNA has also been shown to be affected by post-natal social experiences, stressful environment, and social interaction deprivation. Furthermore, these stimuli have also been linked to increased anxiety, problems with cognition, etc. While a direct link between schizophrenia and BDNF levels hasn't been established, these findings suggest a relation to many problems that are similar to symptoms.[1]
Histone modifications
Histones are proteins that the DNA chromosome are wrapped around. Histones are present as an octamer (set of 8 proteins) and they can be modified through acetylation, methylation, SUMOylation, ubiquitination, phosphorylation, etc. These changes can either open or close up the chromosome. Thus, depending on which histone is modified and the exact process, histone modifications can either silence or promote gene expression (while DNA methylation almost always silences).
Because the sub-field of histone modifications is relatively new, there aren't many results yet. Investigations into these epigenetic regulations indicate that therapeutic pathways targeting epigenetic mechanisms like differential methylation and acetylation may be beneficial.[13] Some studies have found that patients with schizophrenia have higher levels of methylation at H3 (the 3rd histone in the octamer) in the prefrontal cortex, an area that could be related to the negative symptoms. A prominent example is the increased repressive methylation of histone 3 (H3K9me2), which has been associated with both age of disease onset and treatment resistance.[13] It has also been shown that histone acetylation and phosphorylation is increased at the promoter for the BDNF protein, which is involved in learning and memory.[1]
More recent studies have found that postmortem brain tissue from patients with schizophrenia had higher levels of HDAC, histone deacetylase, an enzyme that remove acetyl groups from histones. HDAC1 levels are inversely correlated with GAD67 protein expression, which is decreased in patients with schizophrenia.[12]
Heritable epigenetic alterations
Studies have shown that epigenetic changes can be passed on to future generations through meiosis and mitosis.[16] These findings suggest that environmental factors that the parents face can possibly affect how the child's genetic code is regulated. Research findings have shown this to be true for patients with schizophrenia as well. In rats, the transmission of maternal behavior and even stress responses can be attributed to how certain genes in the hippocampus of the mother are methylated.[1] Another study has shown that the methylation of the BDNF gene, which can be affected by early life stress and abuse, is also transmittable to future generations.[17]
In addition to epigenetic effects as a result of maternal influence during important stages of neurodevelopment, studies show that nutrient deprivation can result in epigenetic modifications that are maintained from generation to generation.[13] Historically, famines are thought to cause changes in epigenetic regulation within the human genome.[13] Specifically, deprivation of nutrients is thought to alter methylation patterns in mammals, and several case studies have shown that periods of famine are positively correlated to increased incidences of schizophrenia in certain populations.[13] Babies born during periods of famine were up to twice as likely to develop schizophrenia or schizophrenia spectrum disorder.[18][19] Thus, researchers believe that the development of schizophrenia is linked to nutrient deprivation. The leading hypothesis for how this is accomplished is via subtle epigenetic alterations following nutrient deprivation, such as the hypermethylation of genes in neurotransmitter pathways, since it is well documented that dietary restriction has an effect on DNA methylation states.[13] This line of thinking is further supported by studies showing that deficiencies in certain nutrients, including choline, folate and vitamin B12 which are required for the creation S-adenosylmethionine (SAM), are also linked with increasing the epigenetic factors associated with increased risk of schizophrenia.[11] Evidence is still mounting in this area, but the existing correlations are notably strong.
Environmental risks and causes
While there haven't been many studies linking environmental factors to schizophrenia-related epigenetics mechanisms at this point in the field, a few studies have shown interesting results. Advanced paternal age is one of the risk factors for schizophrenia, according to recent research. This is through mutagenesis, which cause further spontaneous changes, or through genomic imprinting. As the parent ages, more and more errors may occur in the epigenetic process.[20] There is also evidence of the association between the inhalation of benzene through the burning of wood and schizophrenic development. This might occur through epigenetic changes.[21] Methamphetamine has also been linked to schizophrenia or similar psychotic symptoms. A recent study found that methamphetamine users had altered DNMT1 levels, similar to how patients with schizophrenia have shown abnormal levels of DNMT1 in GABAergic neurons.[22]
Research has shown that there is not a 100% likelihood for genetically inheriting schizophrenia- as in monozygotic twin pairs, when one twin is diagnosed with schizophrenia, there is only a 50% chance that the other twin will also be diagnosed with schizophrenia. This finding shows that environmental influences play a role in the development of schizophrenia.[1]
Maternal effects have also been shown to increase the rate of schizophrenia. It has been hypothesized that babies born in winter and spring have higher rates of schizophrenia than babies born in the summer and fall because of the increase in respiratory infections during the colder months. Furthermore, a study showed that maternal respiratory infection increased the rate of schizophrenia "three- to sevenfold," and if the mothers would not have gotten the respiratory infection, "14 to 21% of schizophrenia cases would have been prevented." The relative amounts of pro-inflammatory and anti-inflammatory compounds found in maternal serum are associated with the onset of schizophrenia, as seen in studies in which amounts of interleukin 6 and interleukin 10, pro- and anti- inflammatory, respectively, were manipulated. It was reported that increasing the amount of interleukin 10 or decreasing the amount of interleukin 6 lessened the effects of the immune system on the fetus.[23] These long-lasting impacts may indicate an epigenetic effects in the offspring, however, it remains unconfirmed. A 2018 study found that patients with schizophrenia had hypomethylation—associated with increased expression of a gene—of the IP6 promoter region compared to the control subjects.[24]
DNA methylation patterns have been linked with increased schizophrenia risk. Specifically, the hypermethylation of the promoter region has been reported to suppress expression of reelin (RELN) in the frontal and prefrontal cortex. Higher DNA methylation levels of RELN promoters has been observed in schizophrenic patients.[25] Several other genes, including GABAergic, dopaminergic, and serotonergic genes, have also been found to have different methylation in patterns in those affected by schizophrenia. Those with schizophrenia have also been noted to have increased amounts of DNMT1, which is involved in the regulation of methylation at CpG sites. Jaffe et al. recently found that 2104 CpG sites were differently methylated in the prefrontal cortex those with schizophrenia as compared to those without schizophrenia. Most notably, the differentially methylated sites were found in genes having to do with "embryonic development, cell fate commitment, and nervous system differentiation" as well as the time period between late gestation and early life.[26]
Prenatal Maternal Stress (PNMS) is also associated with schizophrenia and schizophrenia spectrum disorders (SSD). Several studies have shown that increased PNMS is linked to decreased fetal growth in males who later develop SSD. It has also been found that PNMS leading to increased morbidity and mortality outcomes are also associates with increased risk of SSD development in males. PNMS, especially in early pregnancy, has also been associated to motor deficiencies and behavioral difficulties in the pre-morbid period before schizophrenia onset, most notably in males.[27]
One of the most interesting findings relating an environmental factor with schizophrenic epigenetic mechanisms is exposure to nicotine. It has been widely reported that 80% of patients with schizophrenia use some form of tobacco.[28] Furthermore, smoking appeared to increase cognition in individuals with schizophrenia. However, it was only a recent study Satta et al.that showed that nicotine leads to decreased levels of DNMT1 in GABAergic mouse neurons, a molecule which adds methyl groups to DNA. This led to increased expression of GAD67.[29]
Research limitations
There are several limitations to current research methods and scientific findings. One problem with postmortem studies is that they only demonstrate a single snapshot of a patient with schizophrenia. Thus, it is hard to relate whether biomarker findings are related to the pathology of schizophrenia.
Another limitation is that the most relevant tissue, that of the brain, is impossible to obtain in living, patients with schizophrenia. To work around this, several studies have used more accessible sources, like lymphocytes or germ cell lines, since some studies have shown that epigenetic mutations can be detected in other tissues.
Epigenetic studies of disorders like schizophrenia are also subject to the subjectivity of psychiatric diagnoses and the spectrum-like nature of mental health problems. This problem with classification of mental health problems have led to intermediate phenotypes that might be better fit.[15]
Detection and treatment
The advent of epigenetics as an avenue to pursue schizophrenic research has brought about many possibilities for both early detection, diagnoses, and treatment. While this field is still at an early stage, there have already been promising findings. Some postmortem brain studies looking at the gene expression of histone methylation has shown promising results that might be used for early detection in other patients. However, the bulk of the translational research focus and findings have been on therapeutic interventions.[12]
Therapeutics
Since epigenetic changes are reversible and susceptible pharmacological treatments and drugs, there is a great deal of promise in developing treatments. As many have pointed out, schizophrenia is a lifelong disorder that has widespread effects. Thus, it may not be possible to fully reverse the disease. Although irreversible, the lives of patients with schizophrenia can be greatly improved through treatments that alleviate symptoms. Treatments like anti-psychotic medications for schizophrenia are effective, but they often have serious side effects, and medical practitioners are always looking to improve patients' treatment outcomes. Recent studies indicate that it may be possible to improve upon existing treatments through epigenetic means. This provides a promising new direction for disease management. Pharmaceuticals that directly affect epigenetic markers could be used to improve the efficacy of a patient's current treatment or serve as an updated treatment regiment altogether.
As previously discussed, schizophrenia is associated with elevated levels of gene methylation and repressive histone marks, including H3K9me2. Mood stabilizers have become a therapeutic method of interest in treating schizophrenia due to their ability to reverse epigenetic alterations like repressive histone marks.[30] Mood stabilizers that are known to target epigenetic markers associated with schizophrenia include lithium, valproate, lamotrigine, and carbamazepine.[30]
Targeting histones modifications
HDAC (histone deacetylase) inhibitors are one class of drugs that are being investigated. Studies have shown that levels of reelin and GAD67 (which are decreased in schizophrenic animal models) are both upregulated after treatment with HDAC inhibitors. Furthermore, there is the added benefit of selectivity, as HDAC inhibitors can be specific to cell type, tissue type, and even regions of the brain.[12]
Valproate, a psychotropic drug, is an HDAC inhibitor that is frequently used to treat patients with schizophrenia.[30] Valproate leads to both increased H3 and H4 acetylation as well as increased GAD67 and reelin mRNA levels in lymphocytes.[30] Lithium has also been shown to be highly effective at increasing histone acetylation and presence of GAD67 and reelin transcripts in patients experiencing psychotic symptoms.[30]
HMT (histone demethylase) inhibitors also act on histones. They prevent the demethylation of the H3K4 histone protein and open up that part of the chromatin. Tranylcypromine, an antidepressant, has been shown to have HMT inhibitory properties, and in a study, treatment of patients with schizophrenia with tranylcypromine showed improvements regarding negative symptoms.[12]
In early studies, imipramine, an antidepressant drug, was shown to be able to remove the repressive epigenetic mark H3K9me2.[13] Decreasing repression at this marker appears to improve treatment outcomes for patients taking antipsychotic medications.[31][32]
Targeting DNA methylation
Among long-term antipsychotic users, post-mortem tissue analysis finds that genes that are typically methylated in patients with psychosis are hypomethylated after lifelong antipsychotic use.[33] In other words, anti-psychotic drugs are able to reduce methylation of genes in long-term users. Removing repressive epigenetic marks is only part of how these drugs modify the epigenome of patients with psychotic symptoms. Antipsychotics are associated with both the induction and inhibition of DNA methylation, which can lead to the simultaneous upregulation and downregulation of different genes in patients.[33] While significant variation in methylation patterns have been recorded in patients using antipsychotic drugs, the evidence in this area is not yet sufficient to determine the exact mechanism underlying how these drugs influence methylation.[33] Schizophrenia is a complex, multifaceted illness, and the variety of methylation patterns observed affirms this knowledge.
DNMT inhibitors have also been shown to increase levels of the reeling protein and GAD67 in cell cultures. Some of the current DNMT inhibitors, however, like zebularine and procainamide, do not cross the blood brain barrier and would not prove as effective a treatment. While DNMT inhibitors would prevent the addition of a methyl group, there is also research done on DNA demethylate inducers, which would pharmacologically induce the removal of methyl groups. Current antipsychotic drugs, like clozapine and sulpiride, have been shown to also induce demethylation.[12]
↑Nakamura JP, Schroeder A, Hudson M, Jones N, Gillespie B, Du X, etal. (October 2019). "The maternal immune activation model uncovers a role for the Arx gene in GABAergic dysfunction in schizophrenia". Brain, Behavior, and Immunity. 81: 161–171. doi:10.1016/j.bbi.2019.06.009. PMID31175998. S2CID176295010.
↑Hoek HW, Brown AS, Susser E (August 1998). "The Dutch famine and schizophrenia spectrum disorders". Social Psychiatry and Psychiatric Epidemiology. 33 (8): 373–379. doi:10.1007/s001270050068. PMID9708024. S2CID30257704.
123Lisoway AJ, Chen CC, Zai CC, Tiwari AK, Kennedy JL (June 2021). "Toward personalized medicine in schizophrenia: Genetics and epigenetics of antipsychotic treatment". Schizophrenia Research. 232: 112–124. doi:10.1016/j.schres.2021.05.010. PMID34049235. S2CID235226800.
Further reading
"Epigenetics". Science Online Special Collection. AAAS. October 2010.
Akbarian S (2010). "Epigenetics of schizophrenia". Behavioral Neurobiology of Schizophrenia and Its Treatment. Current Topics in Behavioral Neurosciences. Vol.4. pp.611–628. doi:10.1007/7854_2010_38. ISBN978-3-642-13716-7. PMID21312415.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.