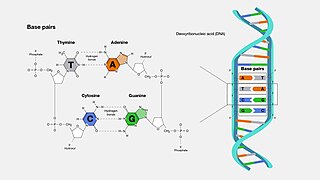
A base pair (bp) is a fundamental unit of double-stranded nucleic acids consisting of two nucleobases bound to each other by hydrogen bonds. They form the building blocks of the DNA double helix and contribute to the folded structure of both DNA and RNA. Dictated by specific hydrogen bonding patterns, "Watson–Crick" base pairs allow the DNA helix to maintain a regular helical structure that is subtly dependent on its nucleotide sequence. The complementary nature of this based-paired structure provides a redundant copy of the genetic information encoded within each strand of DNA. The regular structure and data redundancy provided by the DNA double helix make DNA well suited to the storage of genetic information, while base-pairing between DNA and incoming nucleotides provides the mechanism through which DNA polymerase replicates DNA and RNA polymerase transcribes DNA into RNA. Many DNA-binding proteins can recognize specific base-pairing patterns that identify particular regulatory regions of genes.

Cytosine is one of the four nucleobases found in DNA and RNA, along with adenine, guanine, and thymine. It is a pyrimidine derivative, with a heterocyclic aromatic ring and two substituents attached. The nucleoside of cytosine is cytidine. In Watson-Crick base pairing, it forms three hydrogen bonds with guanine.

In molecular biology, DNA replication is the biological process of producing two identical replicas of DNA from one original DNA molecule. DNA replication occurs in all living organisms acting as the most essential part of biological inheritance. This is essential for cell division during growth and repair of damaged tissues, while it also ensures that each of the new cells receives its own copy of the DNA. The cell possesses the distinctive property of division, which makes replication of DNA essential.

Nucleobases are nitrogen-containing biological compounds that form nucleosides, which, in turn, are components of nucleotides, with all of these monomers constituting the basic building blocks of nucleic acids. The ability of nucleobases to form base pairs and to stack one upon another leads directly to long-chain helical structures such as ribonucleic acid (RNA) and deoxyribonucleic acid (DNA). Five nucleobases—adenine (A), cytosine (C), guanine (G), thymine (T), and uracil (U)—are called primary or canonical. They function as the fundamental units of the genetic code, with the bases A, G, C, and T being found in DNA while A, G, C, and U are found in RNA. Thymine and uracil are distinguished by merely the presence or absence of a methyl group on the fifth carbon (C5) of these heterocyclic six-membered rings. In addition, some viruses have aminoadenine (Z) instead of adenine. It differs in having an extra amine group, creating a more stable bond to thymine.
Deamination is the removal of an amino group from a molecule. Enzymes that catalyse this reaction are called deaminases.

5-Methylcytosine is a methylated form of the DNA base cytosine (C) that regulates gene transcription and takes several other biological roles. When cytosine is methylated, the DNA maintains the same sequence, but the expression of methylated genes can be altered. 5-Methylcytosine is incorporated in the nucleoside 5-methylcytidine.

The CpG sites or CG sites are regions of DNA where a cytosine nucleotide is followed by a guanine nucleotide in the linear sequence of bases along its 5' → 3' direction. CpG sites occur with high frequency in genomic regions called CpG islands.
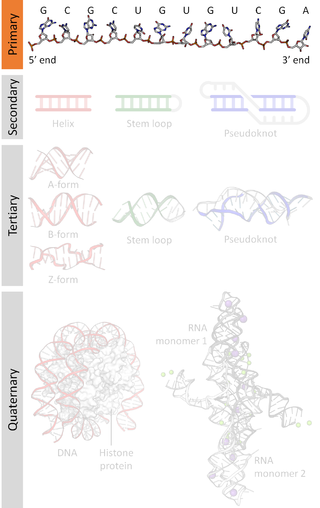
A nucleic acid sequence is a succession of bases within the nucleotides forming alleles within a DNA or RNA (GACU) molecule. This succession is denoted by a series of a set of five different letters that indicate the order of the nucleotides. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, with its double helix, there are two possible directions for the notated sequence; of these two, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.

Molecular genetics is a branch of biology that addresses how differences in the structures or expression of DNA molecules manifests as variation among organisms. Molecular genetics often applies an "investigative approach" to determine the structure and/or function of genes in an organism's genome using genetic screens.

Chargaff's rules state that in the DNA of any species and any organism, the amount of guanine should be equal to the amount of cytosine and the amount of adenine should be equal to the amount of thymine. Further, a 1:1 stoichiometric ratio of purine and pyrimidine bases should exist. This pattern is found in both strands of the DNA. They were discovered by Austrian-born chemist Erwin Chargaff in the late 1940s.

DNA repair is a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encodes its genome. In human cells, both normal metabolic activities and environmental factors such as radiation can cause DNA damage, resulting in tens of thousands of individual molecular lesions per cell per day. Many of these lesions cause structural damage to the DNA molecule and can alter or eliminate the cell's ability to transcribe the gene that the affected DNA encodes. Other lesions induce potentially harmful mutations in the cell's genome, which affect the survival of its daughter cells after it undergoes mitosis. As a consequence, the DNA repair process is constantly active as it responds to damage in the DNA structure. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur, including double-strand breaks and DNA crosslinkages. This can eventually lead to malignant tumors, or cancer as per the two-hit hypothesis.

In molecular biology and genetics, GC-content is the percentage of nitrogenous bases in a DNA or RNA molecule that are either guanine (G) or cytosine (C). This measure indicates the proportion of G and C bases out of an implied four total bases, also including adenine and thymine in DNA and adenine and uracil in RNA.
Nuclear DNA (nDNA), or nuclear deoxyribonucleic acid, is the DNA contained within each cell nucleus of a eukaryotic organism. It encodes for the majority of the genome in eukaryotes, with mitochondrial DNA and plastid DNA coding for the rest. It adheres to Mendelian inheritance, with information coming from two parents, one male and one female—rather than matrilineally as in mitochondrial DNA.
DNA glycosylases are a family of enzymes involved in base excision repair, classified under EC number EC 3.2.2. Base excision repair is the mechanism by which damaged bases in DNA are removed and replaced. DNA glycosylases catalyze the first step of this process. They remove the damaged nitrogenous base while leaving the sugar-phosphate backbone intact, creating an apurinic/apyrimidinic site, commonly referred to as an AP site. This is accomplished by flipping the damaged base out of the double helix followed by cleavage of the N-glycosidic bond.
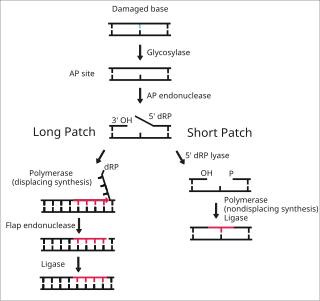
Base excision repair (BER) is a cellular mechanism, studied in the fields of biochemistry and genetics, that repairs damaged DNA throughout the cell cycle. It is responsible primarily for removing small, non-helix-distorting base lesions from the genome. The related nucleotide excision repair pathway repairs bulky helix-distorting lesions. BER is important for removing damaged bases that could otherwise cause mutations by mispairing or lead to breaks in DNA during replication. BER is initiated by DNA glycosylases, which recognize and remove specific damaged or inappropriate bases, forming AP sites. These are then cleaved by an AP endonuclease. The resulting single-strand break can then be processed by either short-patch or long-patch BER.

Activation-induced cytidine deaminase, also known as AICDA, AID and single-stranded DNA cytosine deaminase, is a 24 kDa enzyme which in humans is encoded by the AICDA gene. It creates mutations in DNA by deamination of cytosine base, which turns it into uracil. In other words, it changes a C:G base pair into a U:G mismatch. The cell's DNA replication machinery recognizes the U as a T, and hence C:G is converted to a T:A base pair. During germinal center development of B lymphocytes, AID also generates other types of mutations, such as C:G to A:T. The mechanism by which these other mutations are created is not well understood. It is a member of the APOBEC family.
A postzygotic mutation is a change in an organism's genome that is acquired during its lifespan, instead of being inherited from its parent(s) through fusion of two haploid gametes. Mutations that occur after the zygote has formed can be caused by a variety of sources that fall under two classes: spontaneous mutations and induced mutations. How detrimental a mutation is to an organism is dependent on what the mutation is, where it occurred in the genome and when it occurred.

Pyrimidine dimers are molecular lesions formed from thymine or cytosine bases in DNA via photochemical reactions, commonly associated with direct DNA damage. Ultraviolet light induces the formation of covalent linkages between consecutive bases along the nucleotide chain in the vicinity of their carbon–carbon double bonds. The photo-coupled dimers are fluorescent. The dimerization reaction can also occur among pyrimidine bases in dsRNA —uracil or cytosine. Two common UV products are cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts. These premutagenic lesions alter the structure of the DNA helix and cause non-canonical base pairing. Specifically, adjacent thymines or cytosines in DNA will form a cyclobutane ring when joined together and cause a distortion in the DNA. This distortion prevents replication or transcription machinery beyond the site of the dimerization. Up to 50–100 such reactions per second might occur in a skin cell during exposure to sunlight, but are usually corrected within seconds by photolyase reactivation or nucleotide excision repair. In humans, the most common form of DNA repair is nucleotide excision repair (NER). In contrast, organisms such as bacteria can counterintuitively harvest energy from the sun to fix DNA damage from pyrimidine dimers via photolyase activity. If these lesions are not fixed, polymerase machinery may misread or add in the incorrect nucleotide to the strand. If the damage to the DNA is overwhelming, mutations can arise within the genome of an organism and may lead to the production of cancer cells. Uncorrected lesions can inhibit polymerases, cause misreading during transcription or replication, or lead to arrest of replication. It causes sunburn and it triggers the production of melanin. Pyrimidine dimers are the primary cause of melanomas in humans.
In biochemistry, two biopolymers are antiparallel if they run parallel to each other but with opposite directionality (alignments). An example is the two complementary strands of a DNA double helix, which run in opposite directions alongside each other.
Uracil-DNA glycosylase is an enzyme. Its most important function is to prevent mutagenesis by eliminating uracil from DNA molecules by cleaving the N-glycosidic bond and initiating the base-excision repair (BER) pathway.











