
MEFV is a human gene that provides instructions for making a protein called pyrin. Pyrin is produced in certain white blood cells that play a role in inflammation and in fighting infection. Inside these white blood cells, pyrin is found with the cytoskeleton, the structural framework that helps to define the shape, size, and movement of a cell. Pyrin's protein structure also allows it to interact with other molecules involved in fighting infection and in the inflammatory response.
Periodic fever syndromes are a set of disorders characterized by recurrent episodes of systemic and organ-specific inflammation. Unlike autoimmune disorders such as systemic lupus erythematosus, in which the disease is caused by abnormalities of the adaptive immune system, people with autoinflammatory diseases do not produce autoantibodies or antigen-specific T or B cells. Instead, the autoinflammatory diseases are characterized by errors in the innate immune system.

NLR family pyrin domain containing 3 (NLRP3), is a protein that in humans is encoded by the NLRP3 gene located on the long arm of chromosome 1.

Ubiquitin-activating enzymes, also known as E1 enzymes, catalyze the first step in the ubiquitination reaction, which can target a protein for degradation via a proteasome. This covalent bond of ubiquitin or ubiquitin-like proteins to targeted proteins is a major mechanism for regulating protein function in eukaryotic organisms. Many processes such as cell division, immune responses and embryonic development are also regulated by post-translational modification by ubiquitin and ubiquitin-like proteins.
Nucleotide-binding oligomerization domain-containing protein 2 (NOD2), also known as caspase recruitment domain-containing protein 15 (CARD15) or inflammatory bowel disease protein 1 (IBD1), is a protein that in humans is encoded by the NOD2 gene located on chromosome 16. NOD2 plays an important role in the immune system. It recognizes bacterial molecules (peptidoglycans) and stimulates an immune reaction.

The interleukin-1 receptor antagonist (IL-1RA) is a protein that in humans is encoded by the IL1RN gene.

b(0,+)-type amino acid transporter 1, also known as b(0,+)AT1, is a protein which in humans is encoded by the SLC7A9 gene.

Tumor necrosis factor, alpha-induced protein 3 or A20 is a protein that in humans is encoded by the TNFAIP3 gene.
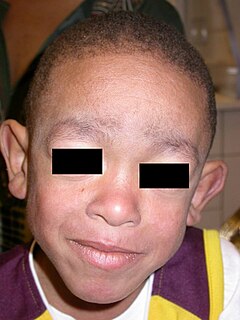
Blau Syndrome is an autosomal dominant genetic inflammatory disorder which affects the skin, eyes, and joints. It is caused by a mutation in the NOD2 (CARD15) gene. Symptoms usually begin before the age of 4, and the disease manifests as early onset cutaneous sarcoidosis, granulomatous arthritis, and uveitis.

PAPA syndrome is an acronym for pyogenic arthritis, pyoderma gangrenosum and acne. It is a rare genetic disorder characterised by its effects on skin and joints.
Cryopyrin-associated periodic syndrome (CAPS) is a group of rare, heterogeneous autoinflammatory disease characterized by interleukin 1β-mediated systemic inflammation and clinical symptoms involving skin, joints, central nervous system, and eyes. It encompasses a spectrum of three clinically overlapping autoinflammatory syndromes including familial cold autoinflammatory syndrome, the Muckle–Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease that were originally thought to be distinct entities, but in fact share a single genetic mutation and pathogenic pathway, and keratoendotheliitis fugax hereditaria in which the autoinflammatory symptoms affect only the anterior segment of the eye.
Deficiency of the interleukin-1–receptor antagonist (DIRA) is an autosomal recessive, genetic autoinflammatory syndrome resulting from mutations in IL1RN, the gene encoding the interleukin 1 receptor antagonist. The mutations result in an abnormal protein that is not secreted, exposing the cells to unopposed interleukin 1 activity. This results in sterile multifocal osteomyelitis, periostitis, and pustulosis due to skin inflammation from birth.

Tetratricopeptide repeat domain 7A (TTC7A) is a protein that in humans is encoded by the TTC7A gene.

Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated Temperature (CANDLE) syndrome is an autosomal recessive disorder that presents itself via various autoinflammatory responses throughout the body, multiple types of skin lesions, and recurrent long-term fever symptoms. The current known cause for the disorder is a mutation in the PSMB8 gene or mutations in other closely related genes. The syndrome was first named and classified in March 2010 after four patients were reviewed with similar symptoms. There have been approximately 30 cases reported in the scientific literature as of 2015.
PLAID syndrome is an inherited condition characterised by antibody deficiency and immune dysregulation, first described in 2012. The name is an acronym of "PLCG2-associated antibody deficiency and immune dysregulation". It is characterised by cold-induced urticaria, autoimmunity, atopy and humoral immune deficiency.
Deficiency of Adenosine deaminase 2 (DADA2) is a monogenic disease associated with systemic inflammation and vasculopathy that affects a wide variety of organs in different patients. As a result, it is hard to characterize a patient with this disorder. Manifestations of the disease include but are not limited to recurrent fever, livedoid rash, various cytopenias, stroke, immunodeficiency, and bone marrow failure. Symptoms often onset during early childhood, but some cases have been discovered as late as 65 years old.
STING-associated vasculopathy with onset in infancy (SAVI) is a rare autoinflammatory vasculopathy associated with the stimulator of interferon genes (STING) protein and characterised by severe skin lesions and interstitial lung disease.
The VEXAS syndrome is an adult-onset autoinflammatory disease affecting males, caused by a mutation in the UBA1 gene. The name derives from Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic.

Aleixo M. Muise is a Canadian scientist, pediatrician and gastroenterologist known for contributions to the understanding of very early onset inflammatory bowel disease. He is a professor of pediatrics at the University of Toronto, a Tier 1 Canada Research Chair in pediatric inflammatory bowel disease, and a pediatric gastroenterologist at the Hospital for Sick Children.
Autoinflammatory diseases (AIDs) are a group of rare disorders caused by a dysfunction of the innate immune system.They are characterised by a perdiodic or chronic systemic inflammation usually without the involvement of adaptive immunity.
