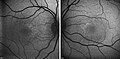
| Hypotrichosis with juvenile macular dystrophy | |
|---|---|
| Other names | Hypotrichosis with juvenile macular degeneration [1] |
 | |
| Hypotrichosis (sparse hair growth) in a 5-year-old boy with HJMD | |
Hypotrichosis with juvenile macular dystrophy (HJMD or CDH3) is an extremely rare congenital disease characterized by sparse hair growth (hypotrichosis) from birth and progressive macular corneal dystrophy.