Haemophilia, or hemophilia, is a mostly inherited genetic disorder that impairs the body's ability to make blood clots, a process needed to stop bleeding. This results in people bleeding for a longer time after an injury, easy bruising, and an increased risk of bleeding inside joints or the brain. Those with a mild case of the disease may have symptoms only after an accident or during surgery. Bleeding into a joint can result in permanent damage while bleeding in the brain can result in long term headaches, seizures, or a decreased level of consciousness.
Haemophilia A is a genetic deficiency in clotting factor VIII, which causes increased bleeding and usually affects males. In the majority of cases it is inherited as an X-linked recessive trait, though there are cases which arise from spontaneous mutations.

Haemophilia B, also spelled hemophilia B, is a blood clotting disorder causing easy bruising and bleeding due to an inherited mutation of the gene for factor IX, and resulting in a deficiency of factor IX. It is less common than factor VIII deficiency.

Coagulation, also known as clotting, is the process by which blood changes from a liquid to a gel, forming a blood clot. It potentially results in hemostasis, the cessation of blood loss from a damaged vessel, followed by repair. The mechanism of coagulation involves activation, adhesion and aggregation of platelets, as well as deposition and maturation of fibrin.

Von Willebrand disease (VWD) is the most common hereditary blood-clotting disorder in humans. An acquired form can sometimes result from other medical conditions. It arises from a deficiency in the quality or quantity of von Willebrand factor (VWF), a multimeric protein that is required for platelet adhesion. It is known to affect several breeds of dogs as well as humans. The three forms of VWD are hereditary, acquired, and pseudo or platelet type. The three types of hereditary VWD are VWD type 1, VWD type 2, and VWD type 3. Type 2 contains various subtypes. Platelet type VWD is also an inherited condition.

Immune thrombocytopenic purpura (ITP), also known as idiopathic thrombocytopenic purpura or immune thrombocytopenia, is a type of thrombocytopenic purpura defined as an isolated low platelet count with a normal bone marrow in the absence of other causes of low platelets. It causes a characteristic red or purple bruise-like rash and an increased tendency to bleed. Two distinct clinical syndromes manifest as an acute condition in children and a chronic condition in adults. The acute form often follows an infection and spontaneously resolves within two months. Chronic immune thrombocytopenia persists longer than six months with a specific cause being unknown.In ITP, your blood does not clot as it should, because you have a low platelet count.

Thrombotic thrombocytopenic purpura (TTP) is a blood disorder that results in blood clots forming in small blood vessels throughout the body. This results in a low platelet count, low red blood cells due to their breakdown, and often kidney, heart, and brain dysfunction. Symptoms may include large bruises, fever, weakness, shortness of breath, confusion, and headache. Repeated episodes may occur.
In biology, hemostasis or haemostasis is a process to prevent and stop bleeding, meaning to keep blood within a damaged blood vessel. It is the first stage of wound healing. This involves coagulation, which changes blood from a liquid to a gel. Intact blood vessels are central to moderating blood's tendency to form clots. The endothelial cells of intact vessels prevent blood clotting with a heparin-like molecule and thrombomodulin, and prevent platelet aggregation with nitric oxide and prostacyclin. When endothelium of a blood vessel is damaged, the endothelial cells stop secretion of coagulation and aggregation inhibitors and instead secrete von Willebrand factor, which initiate the maintenance of hemostasis after injury. Hemostasis involves three major steps:
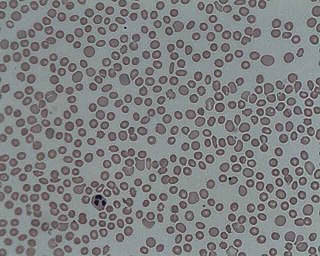
Thrombocytopenia is a condition characterized by abnormally low levels of platelets, also known as thrombocytes, in the blood. Low levels of platelets in turn may lead to prolonged or excessive bleeding. It is the most common coagulation disorder among intensive care patients and is seen in a fifth of medical patients and a third of surgical patients.

Haemophilia C (also known as plasma thromboplastin antecedent deficiency or Rosenthal syndrome) is a mild form of haemophilia affecting both sexes, due to factor XI deficiency. It predominantly occurs in Ashkenazi Jews. It is the fourth most common coagulation disorder after von Willebrand's disease and haemophilia A and B. In the United States, it is thought to affect 1 in 100,000 of the adult population, making it 10% as common as haemophilia A.

Von Willebrand factor (VWF) is a blood glycoprotein involved in hemostasis, specifically, platelet adhesion. It is deficient and/or defective in von Willebrand disease and is involved in many other diseases, including thrombotic thrombocytopenic purpura, Heyde's syndrome, and possibly hemolytic–uremic syndrome. Increased plasma levels in many cardiovascular, neoplastic, metabolic, and connective tissue diseases are presumed to arise from adverse changes to the endothelium, and may predict an increased risk of thrombosis.
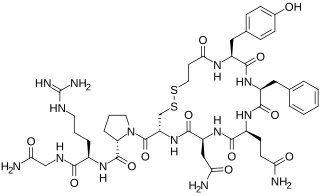
Desmopressin, sold under the trade name DDAVP among others, is a medication used to treat diabetes insipidus, bedwetting, hemophilia A, von Willebrand disease, and high blood urea levels. In hemophilia A and von Willebrand disease, it should only be used for mild to moderate cases. It may be given in the nose, by injection into a vein, by mouth, or under the tongue.

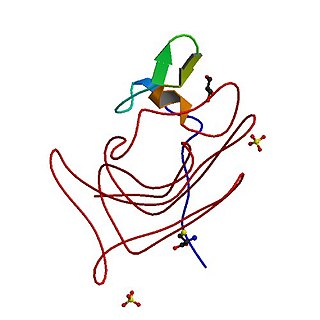
Coagulation factor VII is one of the proteins that causes blood to clot in the coagulation cascade, and in humans is coded for by the gene F7. It is an enzyme of the serine protease class. Once bound to tissue factor released from damaged tissues, it is converted to factor VIIa, which in turn activates factor IX and factor X.

Fresh frozen plasma (FFP) is a blood product made from the liquid portion of whole blood. It is used to treat conditions in which there are low blood clotting factors or low levels of other blood proteins. It may also be used as the replacement fluid in plasma exchange. Using ABO compatible plasma, while not required, may be recommended. Use as a volume expander is not recommended. It is given by slow injection into a vein.
Purpura fulminans is an acute, often fatal, thrombotic disorder which manifests as blood spots, bruising and discolouration of the skin resulting from coagulation in small blood vessels within the skin and rapidly leads to skin necrosis and disseminated intravascular coagulation.

CSL Behring is a biopharmaceutical company, manufacturing plasma-derived, and recombination therapeutic products. Its line of therapies includes products for the treatment of bleeding disorders such as hemophilia); hereditary angioedema; inherited respiratory disease; and neurological disorders. The company's products are also used in cardiac surgery, organ transplantation, burn treatment, and to prevent hemolytic diseases in newborns.
Moroctocog alfa is a recombinant antihemophilic factor genetically engineered from Chinese hamster ovary (CHO) cell line. Chemically it is a glycoprotein. It is manufactured by Genetics Institute, Inc. and used to control and prevent hemorrhagic bleeding and prophylaxis associated with surgery or to reduce the number of spontaneous bleeding episodes in patients with hemophilia A. It is partially a recombinant coagulation factor VIII since it has an amino acid sequence which compares to the 90 + 80 kDa form of factor VIII (BDDrFVIII). It also has posttranslational modifications which are similar to those of the plasma-derived molecule. It can not prevent hemorrhagic bleeding associated with von Willebrand's disease since it is not a von Willebrand factor.
Recombinant factor VIIa also known as eptacog alfa (INN), and sold under the brand name Novoseven among others, is a form of blood factor VII that has been manufactured via recombinant technology. It is administered via an injection into a vein.
Turoctocog alfa is a recombinant antihemophilic factor VIII used for the treatment of and prophylaxis of bleeding patients with haemophilia A. It is marketed by Novo Nordisk. It was approved in the United States, the European Union, and Japan in 2013.
Acquired haemophilia A (AHA) is a rare but potentially life-threatening bleeding disorder characterized by autoantibodies directed against coagulation factor VIII. These autoantibodies constitute the most common spontaneous inhibitor to any coagulation factor and may induce spontaneous bleeding in patients with no previous history of a bleeding disorder.
