
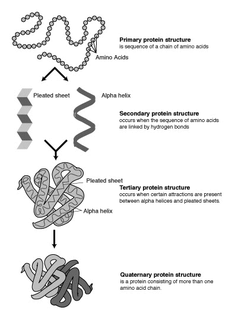
Protein secondary structure is the three dimensional form of local segments of proteins. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.
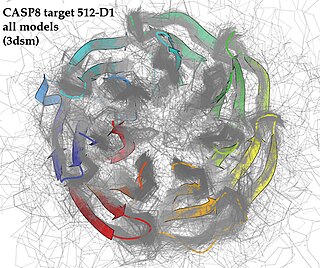
Critical Assessment of protein Structure Prediction (CASP) is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. CASP provides research groups with an opportunity to objectively test their structure prediction methods and delivers an independent assessment of the state of the art in protein structure modeling to the research community and software users. Even though the primary goal of CASP is to help advance the methods of identifying protein three-dimensional structure from its amino acid sequence, many view the experiment more as a “world championship” in this field of science. More than 100 research groups from all over the world participate in CASP on a regular basis and it is not uncommon for entire groups to suspend their other research for months while they focus on getting their servers ready for the experiment and on performing the detailed predictions.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein. Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.
Structural and physical properties of DNA provide important constraints on the binding sites formed on surfaces of DNA-binding proteins. Characteristics of such binding sites may be used for predicting DNA-binding sites from the structural and even sequence properties of unbound proteins. This approach has been successfully implemented for predicting the protein–protein interface. Here, this approach is adopted for predicting DNA-binding sites in DNA-binding proteins. First attempt to use sequence and evolutionary features to predict DNA-binding sites in proteins was made by Ahmad et al. (2004) and Ahmad and Sarai (2005). Some methods use structural information to predict DNA-binding sites and therefore require a three-dimensional structure of the protein, while others use only sequence information and do not require protein structure in order to make a prediction.
Phyre and Phyre2 are free web-based services for protein structure prediction. Phyre is among the most popular methods for protein structure prediction having been cited over 1500 times. Like other remote homology recognition techniques, it is able to regularly generate reliable protein models when other widely used methods such as PSI-BLAST cannot. Phyre2 has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods. Its development is funded by the Biotechnology and Biological Sciences Research Council.
RaptorX is a software and web server for protein structure and function prediction that is free for non-commercial use. RaptorX is among the most popular methods for protein structure prediction. Like other remote homology recognition/protein threading techniques, RaptorX is able to regularly generate reliable protein models when the widely used PSI-BLAST cannot. However, RaptorX is also significantly different from those profile-based methods in that RaptorX excels at modeling of protein sequences without a large number of sequence homologs by exploiting structure information. RaptorX Server has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods.

David Tudor Jones is a Professor of Bioinformatics, and Head of Bioinformatics Group in the University College London. He is also the director in Bloomsbury Center for Bioinformatics, which is a joint Research Centre between UCL and Birkbeck, University of London and which also provides bioinformatics training and support services to biomedical researchers. In 2013, he is a member of editorial boards for PLoS ONE, BioData Mining, Advanced Bioinformatics, Chemical Biology & Drug Design, and Protein: Structure, Function and Bioinformatics.
SWISS-MODEL is a structural bioinformatics web-server dedicated to homology modeling of 3D protein structures. Homology modeling is currently the most accurate method to generate reliable three-dimensional protein structure models and is routinely used in many practical applications. Homology modelling methods make use of experimental protein structures ("templates") to build models for evolutionary related proteins ("targets").
The HH-suite is an open-source software package for sensitive protein sequence searching. It contains programs that can search for similar protein sequences in protein sequence databases. Sequence searches are a standard tool in modern biology with which the function of unknown proteins can be inferred from the functions of proteins with similar sequences. HHsearch and HHblits are two main programs in the package and the entry point to its search function, the latter being a faster iteration. HHpred is an online server for protein structure prediction that uses homology information from HH-suite.
James Andrew Cuff, is a British biophysicist. Cuff has held leadership positions at Harvard University, the Broad Institute, The Wellcome Trust Sanger Institute and the European Bioinformatics Institute.
PredictProtein (PP) is an automatic service that searches up-to-date public sequence databases, creates alignments, and predicts aspects of protein structure and function. Users send a protein sequence and receive a single file with results from database comparisons and prediction methods. PP went online in 1992 at the European Molecular Biology Laboratory; since 1999 it has operated from Columbia University and in 2009 it moved to the Technische Universität München. Although many servers have implemented particular aspects, PP remains the most widely used public server for structure prediction: over 1.5 million requests from users in 104 countries have been handled; over 13000 users submitted 10 or more different queries. PP web pages are mirrored in 17 countries on 4 continents. The system is optimized to meet the demands of experimentalists not experienced in bioinformatics. This implied that we focused on incorporating only high-quality methods, and tried to collate results omitting less reliable or less important ones.

CS23D is a web server to generate 3D structural models from NMR chemical shifts. CS23D combines maximal fragment assembly with chemical shift threading, de novo structure generation, chemical shift-based torsion angle prediction, and chemical shift refinement. CS23D makes use of RefDB and ShiftX.

I-TASSER is a bioinformatics method for predicting three-dimensional structure model of protein molecules from amino acid sequences. It detects structure templates from the Protein Data Bank by a technique called fold recognition. The full-length structure models are constructed by reassembling structural fragments from threading templates using replica exchange Monte Carlo simulations. I-TASSER is one of the most successful protein structure prediction methods in the community-wide CASP experiments.

Coiled-coil domain-containing protein 138, also known as CCDC138, is a human protein encoded by the CCDC138 gene. The exact function of CCDC138 is unknown.
PSI-blast based secondary structure PREDiction (PSIPRED) is a method used to investigate protein structure. It uses artificial neural network machine learning methods in its algorithm. It is a server-side program, featuring a website serving as a front-end interface, which can predict a protein's secondary structure from the primary sequence.
Volume, Area, Dihedral Angle Reporter (VADAR) is a freely available protein structure validation web server that was developed as a collaboration between Dr. Brian Sykes and Dr. David Wishart at the University of Alberta. VADAR consists of >15 different algorithms and programs for assessing and validating peptide and protein structures from their PDB coordinate data. VADAR is capable of determining secondary structure, identifying and classifying six different types of beta turns, determining and calculating the strength of C=O -- N-H hydrogen bonds, calculating residue-specific accessible surface areas (ASA), calculating residue volumes, determining backbone and side chain torsion angles, assessing local structure quality, evaluating global structure quality and identifying residue “outliers”. The results have been validated through extensive comparison to published data and careful visual inspection. VADAR produces both text and graphical output with most of the quantitative data presented in easily viewed tables. In particular, VADAR’s output is presented in a vertical, tabular format with most of the sequence data, residue numbering and any other calculated property or feature presented from top to bottom, rather than from left to right.
ProBiS is a computer software which allows prediction of binding sites and their corresponding ligands for a given protein structure. Initially ProBiS was developed as a ProBiS algorithm by Janez Konc and Dušanka Janežič in 2010 and is now available as ProBiS server, ProBiS CHARMMing server, ProBiS algorithm and ProBiS plugin. The name ProBiS originates from the purpose of the software itself, that is to predict for a given Protein structure Binding Sites and their corresponding ligands.
Coiled-coil domain containing 121 (CCDC121) is a protein encoded by the CCDC121 gene in humans. CCDC121 is located on the minus strand of chromosome 2 and encodes three protein isoforms. All isoforms of CCDC121 contain a domain of unknown function referred to as DUF4515 or pfam14988.