Related Research Articles

Protein secondary structure is the three dimensional form of local segments of proteins. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.
In biology, a sequence motif is a nucleotide or amino-acid sequence pattern that is widespread and usually assumed to be related to biological function of the macromolecule. For example, an N-glycosylation site motif can be defined as Asn, followed by anything but Pro, followed by either Ser or Thr, followed by anything but Pro residue.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.

A protein contact map represents the distance between all possible amino acid residue pairs of a three-dimensional protein structure using a binary two-dimensional matrix. For two residues and , the element of the matrix is 1 if the two residues are closer than a predetermined threshold, and 0 otherwise. Various contact definitions have been proposed: The distance between the Cα-Cα atom with threshold 6-12 Å; distance between Cβ-Cβ atoms with threshold 6-12 Å ; and distance between the side-chain centers of mass.

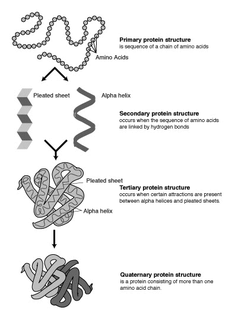
Biomolecular structure is the intricate folded, three-dimensional shape that is formed by a molecule of protein, DNA, or RNA, and that is important to its function. The structure of these molecules may be considered at any of several length scales ranging from the level of individual atoms to the relationships among entire protein subunits. This useful distinction among scales is often expressed as a decomposition of molecular structure into four levels: primary, secondary, tertiary, and quaternary. The scaffold for this multiscale organization of the molecule arises at the secondary level, where the fundamental structural elements are the molecule's various hydrogen bonds. This leads to several recognizable domains of protein structure and nucleic acid structure, including such secondary-structure features as alpha helixes and beta sheets for proteins, and hairpin loops, bulges, and internal loops for nucleic acids. The terms primary, secondary, tertiary, and quaternary structure were introduced by Kaj Ulrik Linderstrøm-Lang in his 1951 Lane Medical Lectures at Stanford University.
Nucleic acid structure prediction is a computational method to determine secondary and tertiary nucleic acid structure from its sequence. Secondary structure can be predicted from one or several nucleic acid sequences. Tertiary structure can be predicted from the sequence, or by comparative modeling.
The Chou–Fasman method is an empirical technique for the prediction of tertiary structures in proteins, originally developed in the 1970s by Peter Y. Chou and Gerald D. Fasman. The method is based on analyses of the relative frequencies of each amino acid in alpha helices, beta sheets, and turns based on known protein structures solved with X-ray crystallography. From these frequencies a set of probability parameters were derived for the appearance of each amino acid in each secondary structure type, and these parameters are used to predict the probability that a given sequence of amino acids would form a helix, a beta strand, or a turn in a protein. The method is at most about 50–60% accurate in identifying correct secondary structures, which is significantly less accurate than the modern machine learning–based techniques.
The GOR method is an information theory-based method for the prediction of secondary structures in proteins. It was developed in the late 1970s shortly after the simpler Chou–Fasman method. Like Chou–Fasman, the GOR method is based on probability parameters derived from empirical studies of known protein tertiary structures solved by X-ray crystallography. However, unlike Chou–Fasman, the GOR method takes into account not only the propensities of individual amino acids to form particular secondary structures, but also the conditional probability of the amino acid to form a secondary structure given that its immediate neighbors have already formed that structure. The method is therefore essentially Bayesian in its analysis.
Secondary structure prediction is a set of techniques in bioinformatics that aim to predict the secondary structures of proteins and nucleic acid sequences based only on knowledge of their primary structure. For proteins, this means predicting the formation of protein structures such as alpha helices and beta strands, while for nucleic acids it means predicting the formation of nucleic acid structures like helixes and stem-loop structures through base pairing and base stacking interactions.
Structural and physical properties of DNA provide important constraints on the binding sites formed on surfaces of DNA-binding proteins. Characteristics of such binding sites may be used for predicting DNA-binding sites from the structural and even sequence properties of unbound proteins. This approach has been successfully implemented for predicting the protein–protein interface. Here, this approach is adopted for predicting DNA-binding sites in DNA-binding proteins. First attempt to use sequence and evolutionary features to predict DNA-binding sites in proteins was made by Ahmad et al. (2004) and Ahmad and Sarai (2005). Some methods use structural information to predict DNA-binding sites and therefore require a three-dimensional structure of the protein, while others use only sequence information and do not require protein structure in order to make a prediction.

Nucleic acid structure refers to the structure of nucleic acids such as DNA and RNA. Chemically speaking, DNA and RNA are very similar. Nucleic acid structure is often divided into four different levels: primary, secondary, tertiary, and quaternary.

Nucleic acid secondary structure is the basepairing interactions within a single nucleic acid polymer or between two polymers. It can be represented as a list of bases which are paired in a nucleic acid molecule. The secondary structures of biological DNAs and RNAs tend to be different: biological DNA mostly exists as fully base paired double helices, while biological RNA is single stranded and often forms complex and intricate base-pairing interactions due to its increased ability to form hydrogen bonds stemming from the extra hydroxyl group in the ribose sugar.

David Tudor Jones is a Professor of Bioinformatics, and Head of Bioinformatics Group in the University College London. He is also the director in Bloomsbury Center for Bioinformatics, which is a joint Research Centre between UCL and Birkbeck, University of London and which also provides bioinformatics training and support services to biomedical researchers. In 2013, he is a member of editorial boards for PLoS ONE, BioData Mining, Advanced Bioinformatics, Chemical Biology & Drug Design, and Protein: Structure, Function and Bioinformatics.
PredictProtein (PP) is an automatic service that searches up-to-date public sequence databases, creates alignments, and predicts aspects of protein structure and function. Users send a protein sequence and receive a single file with results from database comparisons and prediction methods. PP went online in 1992 at the European Molecular Biology Laboratory; since 1999 it has operated from Columbia University and in 2009 it moved to the Technische Universität München. Although many servers have implemented particular aspects, PP remains the most widely used public server for structure prediction: over 1.5 million requests from users in 104 countries have been handled; over 13000 users submitted 10 or more different queries. PP web pages are mirrored in 17 countries on 4 continents. The system is optimized to meet the demands of experimentalists not experienced in bioinformatics. This implied that we focused on incorporating only high-quality methods, and tried to collate results omitting less reliable or less important ones.
Jpred v.4 is the latest version of the JPred Protein Secondary Structure Prediction Server which provides predictions by the JNet algorithm, one of the most accurate methods for secondary structure prediction, that has existed since 1998 in different versions.
The ViennaRNA Package is a set of standalone programs and libraries used for prediction and analysis of RNA secondary structures. The source code for the package is distributed freely and compiled binaries are available for Linux, macOS and Windows platforms. The original paper has been cited over 2000 times.
Machine learning in bioinformatics is the application of machine learning algorithms to bioinformatics, including genomics, proteomics, microarrays, systems biology, evolution, and text mining.
References
- ↑ "Index of /downloads/psipred". bioinfadmin.cs.ucl.ac.uk. Retrieved 26 April 2021.
- ↑ Gajendra P. S. Raghava; Harpreet Kaur. "Prediction of beta turn types" . Retrieved 5 May 2014.
- 1 2 3 Yi-Ping Phoebe Chen (18 January 2005). Bioinformatics Technologies. Springer. p. 107. ISBN 978-3-540-20873-0.
- ↑ Cuff, James A.; Barton, Geoffrey A. (15 August 2000). "Application of multiple sequence alignment profiles to improve protein secondary structure prediction". Proteins. 40 (3): 502–11. doi:10.1002/1097-0134(20000815)40:3<502::aid-prot170>3.0.co;2-q. PMID 10861942.
- ↑ Heringa, Jaap (2000). "Computational Methods for Protein Secondary Structure Prediction Using Multiple Sequence Alignments". Current Protein & Peptide Science. 1 (3): 273–301(29). CiteSeerX 10.1.1.470.7673 . doi:10.2174/1389203003381324. PMID 12369910.
- 1 2 S. C. Rastogi; Namitra Mendiratta; Parag Rastogi (22 May 2013). Bioinformatics: Methods and Applications: (Genomics, Proteomics and Drug Discovery). PHI Learning Pvt. Ltd. pp. 302–. ISBN 978-81-203-4785-4.
- ↑ "PSIPRED | Bioinformatic Technology". 10 April 2014. Retrieved 7 May 2014.
- ↑ "PSIPRED overview" . Retrieved 7 May 2014.
- 1 2 3 4 Jones, David T. (17 September 1999). "Protein Secondary Structure Prediction Based on Position-specific Scoring Matrices" (PDF). Journal of Molecular Biology. 292 (2): 195–202. doi:10.1006/jmbi.1999.3091. PMID 10493868 . Retrieved 7 May 2014.