
Keratitis is a condition in which the eye's cornea, the clear dome on the front surface of the eye, becomes inflamed. The condition is often marked by moderate to intense pain and usually involves any of the following symptoms: pain, impaired eyesight, photophobia, red eye and a 'gritty' sensation. Diagnosis of infectious keratitis is usually made clinically based on the signs and symptoms as well as eye examination, but corneal scrapings may be obtained and evaluated using microbiological culture or other testing to identify the causative pathogen.

Familial Mediterranean fever (FMF) is a hereditary inflammatory disorder. FMF is an autoinflammatory disease caused by mutations in Mediterranean fever gene, which encodes a 781–amino acid protein called pyrin. While all ethnic groups are susceptible to FMF, it usually occurs in people of Mediterranean origin—including Sephardic Jews, Mizrahi Jews, Ashkenazi Jews, Assyrians, Armenians, Azerbaijanis, Druze, Levantines, Kurds, Greeks, Turks and Italians.

Fuchs dystrophy, also referred to as Fuchs endothelial corneal dystrophy (FECD) and Fuchs endothelial dystrophy (FED), is a slowly progressing corneal dystrophy that usually affects both eyes and is slightly more common in women than in men. Although early signs of Fuchs dystrophy are sometimes seen in people in their 30s and 40s, the disease rarely affects vision until people reach their 50s and 60s.

Corneal dystrophy is a group of rare hereditary disorders characterised by bilateral abnormal deposition of substances in the transparent front part of the eye called the cornea.
Cogan syndrome is a rare disorder characterized by recurrent inflammation of the front of the eye and often fever, fatigue, and weight loss, episodes of vertigo (dizziness), tinnitus and hearing loss. It can lead to deafness or blindness if untreated. The classic form of the disease was first described by D. G. Cogan in 1945.
Mucolipidosis type IV is an autosomal recessive lysosomal storage disorder. Individuals with the disorder have many symptoms including delayed psychomotor development and various ocular aberrations. The disorder is caused by mutations in the MCOLN1 gene, which encodes a non-selective cation channel, mucolipin1. These mutations disrupt cellular functions and lead to a neurodevelopmental disorder through an unknown mechanism. Researchers dispute the physiological role of the protein product and which ion it transports.

Muckle–Wells syndrome (MWS) is a rare autosomal dominant disease which causes sensorineural deafness and recurrent hives, and can lead to amyloidosis. Individuals with MWS often have episodic fever, chills, and joint pain. As a result, MWS is considered a type of periodic fever syndrome. MWS is caused by a defect in the CIAS1 gene which creates the protein cryopyrin. MWS is closely related to two other syndromes, familial cold urticaria and neonatal onset multisystem inflammatory disease—in fact, all three are related to mutations in the same gene and subsumed under the term cryopyrin-associated periodic syndromes (CAPS).

NLR family pyrin domain containing 3 (NLRP3), is a protein that in humans is encoded by the NLRP3 gene located on the long arm of chromosome 1.
Lecithin cholesterol acyltransferase deficiency is a disorder of lipoprotein metabolism. The disease has two forms: Familial LCAT deficiency, in which there is complete LCAT deficiency, and Fish-eye disease, in which there is a partial deficiency.
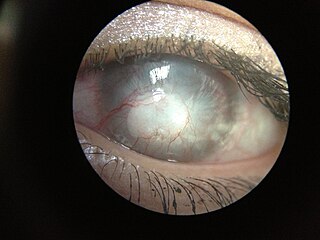
Corneal neovascularization (CNV) is the in-growth of new blood vessels from the pericorneal plexus into avascular corneal tissue as a result of oxygen deprivation. Maintaining avascularity of the corneal stroma is an important aspect of healthy corneal physiology as it is required for corneal transparency and optimal vision. A decrease in corneal transparency causes visual acuity deterioration. Corneal tissue is avascular in nature and the presence of vascularization, which can be deep or superficial, is always pathologically related.

Meesmann corneal dystrophy (MECD) is a rare hereditary autosomal dominant disease that is characterized as a type of corneal dystrophy and a keratin disease. MECD is characterized by the formation of microcysts in the outermost layer of the cornea, known as the anterior corneal epithelium. The anterior corneal epithelium also becomes fragile. This usually affects both eyes rather than a single eye and worsens over time. There are two phenotypes, Meesmann corneal dystrophy 1 (MECD1) and Meesmann corneal dystrophy 2 (MECD2), which affect the genes KRT3 and KRT12, respectively. A heterozygous mutation in either of these genes will lead to a single phenotype. Many with Meesmann corneal dystrophy are asymptomatic or experience mild symptoms.
Cryopyrin-associated periodic syndrome (CAPS) is a group of rare, heterogeneous autoinflammatory disease characterized by interleukin 1β-mediated systemic inflammation and clinical symptoms involving skin, joints, central nervous system, and eyes. It encompasses a spectrum of three clinically overlapping autoinflammatory syndromes including familial cold autoinflammatory syndrome, the Muckle–Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease that were originally thought to be distinct entities, but in fact share a single genetic mutation and pathogenic pathway, and keratoendotheliitis fugax hereditaria in which the autoinflammatory symptoms affect only the anterior segment of the eye.

Posterior polymorphous corneal dystrophy is a type of corneal dystrophy, characterised by changes in Descemet's membrane and endothelial layer. Symptoms mainly consist of decreased vision due to corneal edema. In some cases they are present from birth, other patients are asymptomatic. Histopathological analysis shows that the cells of endothelium have some characteristics of epithelial cells and have become multilayered. The disease was first described in 1916 by Koeppe as keratitis bullosa interna.

NOD-like receptor family pyrin domain containing 11 is a protein that in humans is encoded by the NLRP11 gene located on the long arm of human chromosome 19q13.42. NLRP11 belongs to the NALP subfamily, part of a large subfamily of CATERPILLER. It is also known as NALP11, PYPAF6, NOD17, PAN10, and CLR19.6

Lattice corneal dystrophy type is a rare form of corneal dystrophy. It has no systemic manifestations, unlike the other type of the dystrophy, Lattice corneal dystrophy type II. Lattice corneal dystrophy was first described by Swiss ophthalmologist Hugo Biber in 1890.
NLRP (Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing), also abbreviated as NALP, is a type of NOD-like receptor. NOD-like receptors are a type of pattern recognition receptor that are found in the cytosol of the cell, recognizing signals of antigens in the cell. NLRP proteins are part of the innate immune system and detect conserved pathogen characteristics, or pathogen-associated molecular patterns, such as such as peptidoglycan, which is found on some bacterial cells. It is thought that NLRP proteins sense danger signals linked to microbial products, initiating the processes associated with the activation of the inflammasome, including K+ efflux and caspase 1 activation. NLRPs are also known to be associated with a number of diseases. Research suggests NLRP proteins may be involved in combating retroviruses in gametes. As of now, there are at least 14 different known NLRP genes in humans, which are named NLRP1 through NLRP14. The genes translate into proteins with differing lengths of leucine-rich repeat domains.

Herpetic simplex keratitis is a form of keratitis caused by recurrent herpes simplex virus (HSV) infection in the cornea.

Corneal opacification is a term used when the human cornea loses its transparency. The term corneal opacity is used particularly for the loss of transparency of cornea due to scarring. Transparency of the cornea is dependent on the uniform diameter and the regular spacing and arrangement of the collagen fibrils within the stroma. Alterations in the spacing of collagen fibrils in a variety of conditions including corneal edema, scars, and macular corneal dystrophy is clinically manifested as corneal opacity. The term corneal blindness is commonly used to describe blindness due to corneal opacity.
Exposure keratopathy is medical condition affecting the cornea of eyes. It can lead to corneal ulceration and permanent loss of vision due to corneal opacity.
Peripheral Ulcerative Keratitis (PUK) is a group of destructive inflammatory diseases involving the peripheral cornea in human eyes. The symptoms of PUK include pain, redness of the eyeball, photophobia, and decreased vision accompanied by distinctive signs of crescent-shaped damage of the cornea. The causes of this disease are broad, ranging from injuries, contamination of contact lenses, to association with other systemic conditions. PUK is associated with different ocular and systemic diseases. Mooren's ulcer is a common form of PUK. The majority of PUK is mediated by local or systemic immunological processes, which can lead to inflammation and eventually tissue damage. Standard PUK diagnostic test involves reviewing the medical history and a completing physical examinations. Two major treatments are the use of medications such as corticosteroids or other immunosuppressive agents and surgical resection of the conjunctiva. The prognosis of PUK is unclear with one study providing potential complications. PUK is a rare condition with an estimated incidence of 3 per million annually.
