
Glucocorticoids are a class of corticosteroids, which are a class of steroid hormones. Glucocorticoids are corticosteroids that bind to the glucocorticoid receptor that is present in almost every vertebrate animal cell. The name "glucocorticoid" is a portmanteau and is composed from its role in regulation of glucose metabolism, synthesis in the adrenal cortex, and its steroidal structure.
Mineralocorticoids are a class of corticosteroids, which in turn are a class of steroid hormones. Mineralocorticoids are produced in the adrenal cortex and influence salt and water balances. The primary mineralocorticoid is aldosterone.

A selective progesterone receptor modulator (SPRM) is an agent that acts on the progesterone receptor (PR), the biological target of progestogens like progesterone. A characteristic that distinguishes such substances from full receptor agonists and full antagonists is that their action differs in different tissues, i.e. agonist in some tissues while antagonist in others. This mixed profile of action leads to stimulation or inhibition in tissue-specific manner, which further raises the possibility of dissociating undesirable adverse effects from the development of synthetic PR-modulator drug candidates.
11β-Hydroxysteroid dehydrogenase enzymes catalyze the conversion of inert 11 keto-products (cortisone) to active cortisol, or vice versa, thus regulating the access of glucocorticoids to the steroid receptors.

The glucocorticoid receptor also known as NR3C1 is the receptor to which cortisol and other glucocorticoids bind.

Apparent mineralocorticoid excess is an autosomal recessive disorder causing hypertension, hypernatremia and hypokalemia. It results from mutations in the HSD11B2 gene, which encodes the kidney isozyme of 11β-hydroxysteroid dehydrogenase type 2. In an unaffected individual, this isozyme inactivates circulating cortisol to the less active metabolite cortisone. The inactivating mutation leads to elevated local concentrations of cortisol in the aldosterone sensitive tissues like the kidney. Cortisol at high concentrations can cross-react and activate the mineralocorticoid receptor due to the non-selectivity of the receptor, leading to aldosterone-like effects in the kidney. This is what causes the hypokalemia, hypertension, and hypernatremia associated with the syndrome. Patients often present with severe hypertension and end-organ changes associated with it like left ventricular hypertrophy, retinal, renal and neurological vascular changes along with growth retardation and failure to thrive. In serum both aldosterone and renin levels are low.

The mineralocorticoid receptor, also known as the aldosterone receptor or nuclear receptor subfamily 3, group C, member 2, (NR3C2) is a protein that in humans is encoded by the NR3C2 gene that is located on chromosome 4q31.1-31.2.
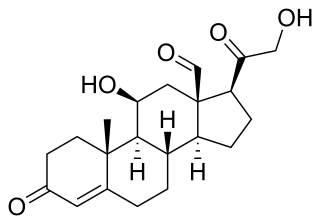
RU-28362 is a synthetic androstane glucocorticoid that was developed by Roussel Uclaf. It is a selective agonist of the glucocorticoid receptor, but not of the mineralocorticoid receptor.

Selective androgen receptor modulators (SARMs) are a class of drugs that selectively activate the androgen receptor in specific tissues, promoting muscle and bone growth while having less effect on male reproductive tissues like the prostate gland.

LGD-2226 is an investigational selective androgen receptor modulator (SARM), which is being developed for treatment of muscle wasting and osteoporosis.
Enobosarm, also formerly known as ostarine and by the developmental code names GTx-024, MK-2866, and S-22, is a selective androgen receptor modulator (SARM) which is under development for the treatment of androgen receptor-positive breast cancer in women and for improvement of body composition in people taking GLP-1 receptor agonists like semaglutide. It was also under development for a variety of other indications, including treatment of cachexia, Duchenne muscular dystrophy, muscle atrophy or sarcopenia, and stress urinary incontinence, but development for all other uses has been discontinued. Enobosarm was evaluated for the treatment of muscle wasting related to cancer in late-stage clinical trials, and the drug improved lean body mass in these trials, but it was not effective in improving muscle strength. As a result, enobosarm was not approved and development for this use was terminated. Enobosarm is taken by mouth.

Selective glucocorticoid receptor modulators (SEGRMs) and selective glucocorticoid receptor agonists (SEGRAs) formerly known as dissociated glucocorticoid receptor agonists (DIGRAs) are a class of experimental drugs designed to share many of the desirable anti-inflammatory, immunosuppressive, or anticancer properties of classical glucocorticoid drugs but with fewer side effects such as skin atrophy. Although preclinical evidence on SEGRAMs’ anti-inflammatory effects are culminating, currently, the efficacy of these SEGRAMs on cancer are largely unknown.

Samidorphan is an opioid antagonist that in the form of olanzapine/samidorphan is used in the treatment of schizophrenia and bipolar disorder. Samidorphan reduces the weight gain associated with olanzapine. Samidorphan is taken by mouth.

Mexrenone is a steroidal antimineralocorticoid of the spirolactone group related to spironolactone that was never marketed. It is the lactonic form of mexrenoic acid (mexrenoate), and mexrenoate potassium (SC-26714), the potassium salt of mexrenoic acid, also exists. In addition to the mineralocorticoid receptor, mexrenone also binds to the glucocorticoid, androgen, and progesterone receptors. Relative to spironolactone, it has markedly reduced antiandrogen activity. Eplerenone is the 9-11α-epoxy analogue of mexrenone.
The corticosteroid receptors are receptors for corticosteroids. Corticosteroid receptors mediate the target organ response to the major products of the adrenal cortex, glucocorticoids, and mineralocorticoids. They are members of the intracellular receptor superfamily which is highly evolutionarily conserved, and includes receptors for thyroid hormones, vitamin D, sex steroids, and retinoids. They include the following two nuclear receptors:
Membrane glucocorticoid receptors (mGRs) are a group of receptors which bind and are activated by glucocorticoids such as cortisol and corticosterone, as well as certain exogenous glucocorticoids such as dexamethasone. Unlike the classical nuclear glucocorticoid receptor (GR), which mediates its effects via genomic mechanisms, mGRs are cell surface receptors which rapidly alter cell signaling via modulation of intracellular signaling cascades. The identities of the mGRs have yet to be fully elucidated, but are thought to include membrane-associated classical GRs as well as yet-to-be-characterized G protein-coupled receptors (GPCRs). Rapid effects of dexamethasone were found not be reversed by the GR antagonist mifepristone, indicating additional receptors besides just the classical GR.
Membrane mineralocorticoid receptors (mMRs) or membrane aldosterone receptors are a group of receptors which bind and are activated by mineralocorticoids such as aldosterone. Unlike the classical nuclear mineralocorticoid receptor (MR), which mediates its effects via genomic mechanisms, mMRs are cell surface receptors which rapidly alter cell signaling via modulation of intracellular signaling cascades. The identities of the mMRs have yet to be fully elucidated, but are thought to include membrane-associated classical MRs as well as yet-to-be-characterized G protein-coupled receptors (GPCRs). Rapid effects of aldosterone were found not be reversed by the MR antagonist spironolactone, indicating additional receptors besides just the classical MR. It has been estimated that as much as 50% of the rapid actions of aldosterone are mediated by mMRs that are not the classical MR, based on findings of insensitivity to classical mR antagonists.
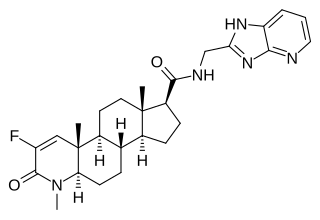
MK-0773, also known as PF-05314882, is a steroidal, orally active selective androgen receptor modulator (SARM) that was under development by Merck and GTx for the treatment of sarcopenia in women and men. Clinical trials for sarcopenia began in late 2007 but the collaboration between Merck and GTx ended in early 2010 and GTx terminated development of MK-0773 shortly thereafter. MK-0773 was developed as a more advanced version of the related compound TFM-4AS-1.

RU-59063 is a nonsteroidal androgen or selective androgen receptor modulator (SARM) which was first described in 1994 and was never marketed. It was originally thought to be a potent antiandrogen, but subsequent research found that it actually possesses dose-dependent androgenic activity, albeit with lower efficacy than dihydrotestosterone (DHT). The drug is an N-substituted arylthiohydantoin and was derived from the first-generation nonsteroidal antiandrogen (NSAA) nilutamide. The second-generation NSAAs enzalutamide, RD-162, and apalutamide were derived from RU-59063.
Generalized glucocorticoid resistance or Chrousos syndrome is a rare genetic disorder that can run in families or be sporadic. It is characterized by partial or generalized target-tissue insensitivity to glucocorticoids.
