In organic chemistry, an imine is a functional group or organic compound containing a carbon–nitrogen double bond. The nitrogen atom can be attached to a hydrogen or an organic group (R). The carbon atom has two additional single bonds. Imines are common in synthetic and naturally occurring compounds and they participate in many reactions.
In organic chemistry, the Mannich reaction is a three-component organic reaction that involves the amino alkylation of an acidic proton next to a carbonyl functional group by formaldehyde and a primary or secondary amine or ammonia. The final product is a β-amino-carbonyl compound also known as a Mannich base. Reactions between aldimines and α-methylene carbonyls are also considered Mannich reactions because these imines form between amines and aldehydes. The reaction is named after Carl Mannich.
Reductive amination is a form of amination that converts a carbonyl group to an amine via an intermediate imine. The carbonyl group is most commonly a ketone or an aldehyde. It is a common method to make amines and is widely used in green chemistry since it can be done catalytically in one-pot under mild conditions. In biochemistry, dehydrogenase enzymes use reductive amination to produce the amino acid glutamate. Additionally, there is ongoing research on alternative synthesis mechanisms with various metal catalysts which allow the reaction to be less energy taxing, and require milder reaction conditions. Investigation into biocatalysts, such as imine reductases, have allowed for higher selectivity in the reduction of chiral amines which is an important factor in pharmaceutical synthesis.
The Reformatsky reaction is an organic reaction which condenses aldehydes or ketones with α-halo esters using metallic zinc to form β-hydroxy-esters:

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.
The Strecker amino acid synthesis, also known simply as the Strecker synthesis, is a method for the synthesis of amino acids by the reaction of an aldehyde with cyanide in the presence of ammonia. The condensation reaction yields an α-aminonitrile, which is subsequently hydrolyzed to give the desired amino acid. The method is used for the commercial production of racemic methionine from methional.
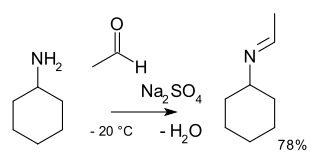
In organic chemistry, alkylimino-de-oxo-bisubstitution is the organic reaction of carbonyl compounds with amines to imines. The reaction name is based on the IUPAC Nomenclature for Transformations. The reaction is acid catalyzed and the reaction type is nucleophilic addition of the amine to the carbonyl compound followed by transfer of a proton from nitrogen to oxygen to a stable hemiaminal or carbinolamine. With primary amines, water is lost in an elimination reaction to an imine. With aryl amines, especially stable Schiff bases are formed.
The Weinreb ketone synthesis or Weinreb–Nahm ketone synthesis is a chemical reaction used in organic chemistry to make carbon–carbon bonds. It was discovered in 1981 by Steven M. Weinreb and Steven Nahm as a method to synthesize ketones. The original reaction involved two subsequent substitutions: the conversion of an acid chloride with N,O-Dimethylhydroxylamine, to form a Weinreb–Nahm amide, and subsequent treatment of this species with an organometallic reagent such as a Grignard reagent or organolithium reagent. Nahm and Weinreb also reported the synthesis of aldehydes by reduction of the amide with an excess of lithium aluminum hydride.

In organic chemistry, aziridines are organic compounds containing the aziridine functional group, a three-membered heterocycle with one amine and two methylene bridges. The parent compound is aziridine, with molecular formula C2H4NH. Several drugs feature aziridine rings, including mitomycin C, porfiromycin, and azinomycin B (carzinophilin).
In organophosphorus chemistry, the Kabachnik–Fields reaction is a three-component organic reaction forming α-aminomethylphosphonates from an amine, a carbonyl compound, and a dialkyl phosphonate, (RO)2P(O)H (that are also called dialkylphosphites). Aminophosphonates are synthetic targets of some importance as phosphorus analogues of α-amino acids (a bioisostere). This multicomponent reaction was independently discovered by Martin Kabachnik and Ellis K. Fields in 1952. The reaction is very similar to the two-component Pudovik reaction, which involves condensation of the phosphite and a preformed imine.
The Abramov reaction is the related conversions of trialkyl to α-hydroxy phosphonates by the addition to carbonyl compounds. In terms of mechanism, the reaction involves attack of the nucleophilic phosphorus atom on the carbonyl carbon. It was named after the Russian chemist Vasilii Semenovich Abramov (1904–1968) in 1957.

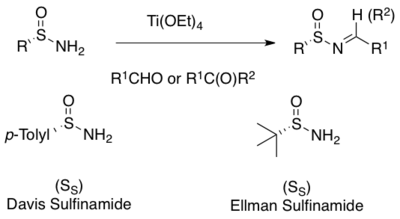
tert-Butanesulfinamide is an organosulfur compound and a member of the class of sulfinamides. Both enantiomeric forms are commercially available and are used in asymmetric synthesis as chiral auxiliaries, often as chiral ammonia equivalents for the synthesis of amines. tert-Butanesulfinamide and the associated synthetic methodology was introduced in 1997 by Jonathan A. Ellman et al.
In organosulfur chemistry, sulfinamide is a functional group with the structure R−S(O)−NR2. This functionality is composed of a sulfur-carbon single bond, a sulfur-nitrogen single bond, and a sulfur-oxygen (S-O) bond. As a non-bonding electron pair is present on the sulfur, the sulfur atom is a stable stereogenic centre, and so these compounds are chiral. They are sometimes referred to as S-chiral sulfinamides. Sulfinamides are amides of sulfinic acid.

An oxaziridine is an organic molecule that features a three-membered heterocycle containing oxygen, nitrogen, and carbon. In their largest application, oxaziridines are intermediates in the industrial production of hydrazine. Oxaziridine derivatives are also used as specialized reagents in organic chemistry for a variety of oxidations, including alpha hydroxylation of enolates, epoxidation and aziridination of olefins, and other heteroatom transfer reactions. Oxaziridines also serve as precursors to nitrones and participate in [3+2] cycloadditions with various heterocumulenes to form substituted five-membered heterocycles. Chiral oxaziridine derivatives effect asymmetric oxygen transfer to prochiral enolates as well as other substrates. Some oxaziridines also have the property of a high barrier to inversion of the nitrogen, allowing for the possibility of chirality at the nitrogen center.
Franklin Arnold Davis is the Laura H. Carnell Professor of Chemistry at Temple University in Philadelphia, Pennsylvania. He is most notable for his development of sulfur-nitrogen reagents including N-sulfonyloxaziridine for oxidations and asymmetric hydroxylations and N-sulfinyl imines for the asymmetric synthesis of chiral amine derivatives. The reagents are commonly called Davis oxaziridines and Davis sulfinamides, respectively. Davis oxidation and Davis' reagent are both named after him.
In organic chemistry, the Roskamp reaction is a name reaction describing the reaction between α-diazoesters (such as ethyl diazoacetate) and aldehydes to form β-ketoesters, often utilizing various Lewis acids (such as BF3, SnCl2, and GeCl2) as catalysts. The reaction is notable for its mild reaction conditions and selectivity.
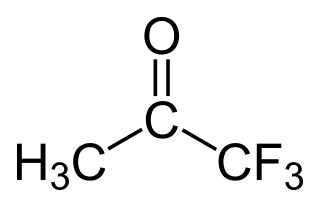
Trifluoroacetone (1,1,1-trifluoroacetone) is an organofluorine compound with the chemical formula CF3C(O)CH3. The compound is a colorless liquid with chloroform-like odour.
In organic chemistry, the Davis oxidation or Davis' oxaziridine oxidation refers to oxidations involving the use of the Davis reagent or other similar oxaziridine reagents. This reaction mainly refers to the generation of α-hydroxy carbonyl compounds (acyloins) from ketones or esters. The reaction is carried out in a basic environment to generate the corresponding enolate from the ketone or ester. This reaction has been shown to work for amides.
The De Kimpe aziridine synthesis is a name reaction of organic chemistry, for the generation of aziridines by the reaction of α-chloroimines with nucleophiles such as hydride, cyanide, or Grignard reagents.
The ketimine Mannich reaction is an asymmetric synthetic technique using differences in starting material to push a Mannich reaction to create an enantiomeric product with steric and electronic effects, through the creation of a ketimine group. Typically, this is done with a reaction with proline or another nitrogen-containing heterocycle, which control chirality with that of the catalyst. This has been theorized to be caused by the restriction of undesired (E)-isomer by preventing the ketone from accessing non-reactive tautomers. Generally, a Mannich reaction is the combination of an amine, a ketone with a β-acidic proton and aldehyde to create a condensed product in a β-addition to the ketone. This occurs through an attack on the ketone with a suitable catalytic-amine unto its electron-starved carbon, from which an imine is created. This then undergoes electrophilic addition with a compound containing an acidic proton. It is theoretically possible for either of the carbonyl-containing molecules to create diastereomers, but with the addition of catalysts which restrict addition as of the enamine creation, it is possible to extract a single product with limited purification steps and in some cases as reported by List et al.; practical one-pot syntheses are possible. The process of selecting a carbonyl-group gives the reaction a direct versus indirect distinction, wherein the latter case represents pre-formed products restricting the reaction's pathway and the other does not. Ketimines selects a reaction group, and circumvent a requirement for indirect pathways.