
Joubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.

Gardner's syndrome is a subtype of familial adenomatous polyposis (FAP). Gardner syndrome is an autosomal dominant form of polyposis characterized by the presence of multiple polyps in the colon together with tumors outside the colon. The extracolonic tumors may include osteomas of the skull, thyroid cancer, epidermoid cysts, fibromas, as well as the occurrence of desmoid tumors in approximately 15% of affected individuals.
Jacobsen syndrome is a rare chromosomal disorder resulting from deletion of genes from chromosome 11 that includes band 11q24.1. It is a congenital disorder. Since the deletion takes place on the q arm of chromosome 11, it is also called 11q terminal deletion disorder. The deletion may range from 5 million to 16 million deleted DNA base pairs. The severity of symptoms depends on the number of deletions; the more deletions there are, the more severe the symptoms are likely to be.
Van der Woude syndrome (VDWS) is a genetic disorder characterized by the combination of lower lip pits, cleft lip with or without cleft palate (CL/P), and cleft palate only (CPO). The frequency of orofacial clefts ranges from 1:1000 to 1:500 births worldwide, and there are more than 400 syndromes that involve CL/P. VWS is distinct from other clefting syndromes due to the combination of cleft lip and palate (CLP) and CPO within the same family. Other features frequently associated with VWS include hypodontia in 10-81% of cases, narrow arched palate, congenital heart disease, heart murmur and cerebral abnormalities, syndactyly of the hands, polythelia, ankyloglossia, and adhesions between the upper and lower gum pads.

Bannayan–Riley–Ruvalcaba syndrome (BRRS) is a rare overgrowth syndrome and hamartomatous disorder with occurrence of multiple subcutaneous lipomas, macrocephaly and hemangiomas. The disease is inherited in an autosomal dominant manner. The disease belongs to a family of hamartomatous polyposis syndromes, which also includes Peutz–Jeghers syndrome, juvenile polyposis and Cowden syndrome. Mutation of the PTEN gene underlies this syndrome, as well as Cowden syndrome, Proteus syndrome, and Proteus-like syndrome, these four syndromes are referred to as PTEN Hamartoma-Tumor Syndromes.
Ectrodactyly–ectodermal dysplasia–cleft syndrome, or EEC, and also referred to as EEC syndrome and split hand–split foot–ectodermal dysplasia–cleft syndrome is a rare form of ectodermal dysplasia, an autosomal dominant disorder inherited as a genetic trait. EEC is characterized by the triad of ectrodactyly, ectodermal dysplasia, and facial clefts. Other features noted in association with EEC include vesicoureteral reflux, recurrent urinary tract infections, obstruction of the nasolacrimal duct, decreased pigmentation of the hair and skin, missing or abnormal teeth, enamel hypoplasia, absent punctae in the lower eyelids, photophobia, occasional cognitive impairment and kidney anomalies, and conductive hearing loss.

1p36 deletion syndrome is a congenital genetic disorder characterized by moderate to severe intellectual disability, delayed growth, hypotonia, seizures, limited speech ability, malformations, hearing and vision impairment, and distinct facial features. The symptoms may vary, depending on the exact location of the chromosomal deletion.
Rosselli–Gulienetti syndrome, also known as Zlotogora–Ogur syndrome and Bowen–Armstrong syndrome, is a type of congenital ectodermal dysplasia syndrome. The syndrome is relatively rare and has only been described in a few cases.
Young–Simpson syndrome (YSS) is a rare congenital disorder with symptoms including hypothyroidism, heart defects, facial dysmorphism, cryptorchidism in males, hypotonia, intellectual disability, and postnatal growth retardation.

Frontonasal dysplasia (FND) is a congenital malformation of the midface. For the diagnosis of FND, a patient should present at least two of the following characteristics: hypertelorism, a wide nasal root, vertical midline cleft of the nose and/or upper lip, cleft of the wings of the nose, malformed nasal tip, encephalocele or V-shaped hair pattern on the forehead. The cause of FND remains unknown. FND seems to be sporadic (random) and multiple environmental factors are suggested as possible causes for the syndrome. However, in some families multiple cases of FND were reported, which suggests a genetic cause of FND.

Orofaciodigital syndrome 1 (OFD1), also called Papillon-League and Psaume syndrome, is an X-linked congenital disorder characterized by malformations of the face, oral cavity, and digits with polycystic kidney disease and variable involvement of the central nervous system.
A facial cleft is an opening or gap in the face, or a malformation of a part of the face. Facial clefts is a collective term for all sorts of clefts. All structures like bone, soft tissue, skin etc. can be affected. Facial clefts are extremely rare congenital anomalies. There are many variations of a type of clefting and classifications are needed to describe and classify all types of clefting. Facial clefts hardly ever occur isolated; most of the time there is an overlap of adjacent facial clefts.
Malpuech facial clefting syndrome, also called Malpuech syndrome or Gypsy type facial clefting syndrome, is a rare congenital syndrome. It is characterized by facial clefting, a caudal appendage, growth deficiency, intellectual and developmental disability, and abnormalities of the renal system (kidneys) and the male genitalia. Abnormalities of the heart, and other skeletal malformations may also be present. The syndrome was initially described by Georges Malpuech and associates in 1983. It is thought to be genetically related to Juberg-Hayward syndrome. Malpuech syndrome has also been considered as part of a spectrum of congenital genetic disorders associated with similar facial, urogenital and skeletal anomalies. Termed "3MC syndrome", this proposed spectrum includes Malpuech, Michels and Mingarelli-Carnevale (OSA) syndromes. Mutations in the COLLEC11 and MASP1 genes are believed to be a cause of these syndromes. The incidence of Malpuech syndrome is unknown. The pattern of inheritance is autosomal recessive, which means a defective (mutated) gene associated with the syndrome is located on an autosome, and the syndrome occurs when two copies of this defective gene are inherited.
Legius syndrome (LS) is an autosomal dominant condition characterized by cafe au lait spots. It was first described in 2007 and is often mistaken for neurofibromatosis type I. It is caused by mutations in the SPRED1 gene. It is also known as neurofibromatosis type 1-like syndrome.
Goldberg–Shprintzen is a very rare connective tissue condition associated with mutations in KIAA1279 gene which encodes KIF-binding protein (KBP), a protein that may interact with microtubules and actin filaments. KBP may play a key role in cytoskeleton formation and neurite growth.
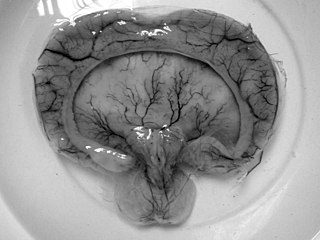
Young–Madders syndrome, alternatively known as Pseudotrisomy 13 syndrome or holoprosencephaly–polydactyly syndrome, is a genetic disorder resulting from defective and duplicated chromosomes which result in holoprosencephaly, polydactyly, facial malformations and intellectual disability, with a significant variance in the severity of symptoms being seen across known cases. Many cases often suffer with several other genetic disorders, and some have presented with hypoplasia, cleft lip, cardiac lesions and other heart defects. In one case in 1991 and another in 2000 the condition was found in siblings who were the product of incest. Many cases are diagnosed prenatally and often in siblings. Cases are almost fatal in the prenatal stage with babies being stillborn.
Fryns-Aftimos syndrome is a rare chromosomal condition and is associated with pachygyria, severe mental retardation, epilepsy and characteristic facial features. This syndrome is a malformation syndrome, characterized by numerous facial dysmorphias not limited to hypertelorism, iris or retinal coloboma, cleft lip, and congenital heart defects. This syndrome has been seen in 30 unrelated people. Characterized by a de novo mutation located on chromosome 7p22, there is typically no family history prior to onset. The severity of the disorder can be determined by the size of the deletion on 7p22, enveloping the ACTB gene and surrounding genes, which is consistent with a contiguous gene deletion syndrome. Confirming a diagnosis of Fryns-Aftimos syndrome typically consists of serial single-gene testing or multigene panel of genes of interest or exome sequencing.
Kahrizi syndrome (KHRZ) is an autosomal-recessive disease that is identified by mental retardation, cataracts, coloboma, kyphosis, and coarse facial features caused by a homozygous mutation in the SRD5A3 gene.

A bifid nose is an uncommon congenital malformation which is characterized by the presence of a cleft between the two nostrils of the nose. It is the result of a disturbance during embryological nose development.
Holoprosencephaly-ectrodactyly-cleft lip/palate syndrome, also simply known as Hartsfield syndrome, is a rare genetic disorder characterized by the presence of variable holoprosencephaly, ectrodactyly, cleft lip and palate, alongside generalized ectodermal abnormalities. Additional findings include endocrine anomalies and developmental delays.
